正常な糖質バランスと血糖値の維持編集
肝臓や腎臓のグリコーゲンは、血糖値を維持するために、素早くアクセスできる貯蔵グルコースの形態として機能する。 炭水化物を含む食事の後約3時間は、高いインスリンレベルが肝細胞に、血液からグルコースを取り込み、グルコキナーゼという酵素でグルコース-6-リン酸(G6P)に変換し、G6P分子をグリコーゲン鎖の末端に加えるよう指令する(グリコーゲン合成)。
食事の消化が完了すると、インスリン濃度が低下し、肝細胞の酵素系がグリコーゲンの鎖からグルコース分子をG6Pの形で除去し始める。 この過程はグリコーゲン分解と呼ばれる。 G6Pは、グルコース-6-ホスファターゼによってリン酸が切断されない限り、肝細胞内に留まる。 この脱リン酸化反応により、遊離グルコースと遊離PO
4アニオンが生成される。 遊離グルコース分子は肝細胞から血液中に輸送され、脳や他の臓器への十分なグルコース供給を維持することができる。 グリコーゲン分解は、成人の体が必要とするグルコースを12~18時間供給できる。
空腹状態が数時間以上続くと、インスリンレベルの低下により、筋肉タンパク質や脂肪組織からのトリグリセリドが異化されるようになる。 これらのプロセスの産物は、アミノ酸(主にアラニン)、遊離脂肪酸、および乳酸である。 トリグリセリドからの遊離脂肪酸は、ケトンとアセチル-CoAに変換される。 アミノ酸と乳酸は、肝細胞内で糖新生という過程を経て、新たにG6Pを合成するために使われる。 通常の糖新生の最後のステップは、グリコーゲン分解の最後のステップと同様に、グルコース-6-ホスファターゼによるG6Pの脱リン酸化で遊離グルコースとPO
したがって、グルコース-6-ホスファターゼは空腹時のグルコース生成の二つの主要プロセスの両方の最後の、重要な、ステップを仲介しているのである。
病態生理編集
グルコース-6-ホスファターゼの欠損による主要な代謝作用は、低血糖、乳酸アシドーシス、高トリグリセリド血症、および高尿酸血症である。

GSD Iの低血糖は「空腹」あるいは「吸収後」と呼ばれ、通常食事の完全消化後4時間程度で起こります。 空腹時に十分な血糖値を維持できないのは、グリコーゲン分解と糖新生の両方が複合的に障害されているためである。 空腹時低血糖は、しばしばGSD Iにおける最も重要な問題であり、典型的には診断に至る問題である。 慢性的な低血糖は、慢性的に低いインスリンレベル、高いグルカゴンおよびコルチゾールレベルなどの二次的な代謝適応を引き起こす。 乳酸は肝臓と筋肉の両方で生成され、NAD+によってピルビン酸に酸化された後、糖新生経路を経由してG6Pに変換される。 G6Pが蓄積されると乳酸からピルビン酸への変換が阻害される。 空腹時にはグルコースが低下するため、乳酸値が上昇する。 GSD Iの患者では、正常なグルコースレベルが回復しても、乳酸値は完全に正常値まで下がらない。
トリグリセリド産生の増幅による高トリグリセリド血症は、慢性的に低いインスリンレベルによって増幅される、糖新生の障害の別の間接的な影響である。 空腹時には、トリグリセリドの遊離脂肪酸、ケトン体、そして最終的にはアセチルCoAへの正常な変換が損なわれる。 高尿酸血症は、増加したG6Pがペントースリン酸経路で代謝される際に生成される尿酸の生成量増加と排泄量の減少の組み合わせから生じる。 また、プリン体の分解による副産物でもある。 尿酸は、乳酸や他の有機酸と競合して、尿中に排泄される。 GSDでは、ペントースリン酸経路でのG6Pの利用可能性の増加、異化率の増加、高濃度の乳酸による尿中排泄の減少が組み合わさって、尿酸値が通常の数倍となる。 高尿酸血症は何年も無症状であるが、腎臓や関節に徐々に障害が生じる。
乳酸値の上昇と乳酸アシドーシス編集
GSD Iのすべての患者で、糖新生の障害により血中の乳酸レベルが高いことが観察される。 ベースラインの上昇は一般に4~10mol/mLであり、臨床的な影響は生じない。 しかし、低血糖が続くと、乳酸値は突然上昇し、乳酸アシドーシスの閾値である15mol/mLを超える。 乳酸アシドーシスの症状には嘔吐と過呼吸があり、いずれもGSD Iの設定における低血糖を悪化させる。急性乳酸アシドーシスの場合、患者は血液酸素を安定させ、血糖を回復するための緊急治療が必要となる。 最初の症状は一般的に嘔吐と脱水であり、いずれも胃腸炎や肺炎などの小児感染症を模倣するため、未診断の小児における乳酸アシドーシスを適切に特定することは困難である。 さらに、これらの一般的な感染症はいずれも未診断の小児においてより重度の低血糖を誘発することがあり、根本的な原因の診断を困難にしている。 成人および小児では、高濃度の乳酸が筋肉に大きな不快感を与える。 この不快感は、ランナーがスプリント後に大腿四頭筋に感じる灼熱感を増幅したもので、乳酸が短時間蓄積されることによって生じるものである。
尿酸値の上昇と合併症
高尿酸値は、GSD I 患者における乳酸値上昇の結果としてしばしば認められる。 乳酸値が上昇すると、血液中の乳酸が尿酸塩と同じ腎尿細管輸送機構で競合し、腎臓から尿中に排出される尿酸塩の速度が制限される。 また、プリン体の異化作用が亢進している場合は、さらなる要因になります。 尿酸値は、適切な治療が行われない場合、6〜12mg/dl(530〜1060umol/L)がGSD Iの患者さんでは一般的である。 一部の患者では、血中尿酸値を下げるためにアロプリノールという薬剤を使用する必要があります。 GSD I 患者の高尿酸血症の結果、腎臓結石の発生や関節への尿酸結晶の蓄積が起こり、それぞれ腎臓病や痛風につながる。
高脂血症と血漿効果編集
GSD I におけるトリグリセリド上昇は、長引く低血糖が頻繁に起こる患者の血清インスリン低下から生じるものです。 また、細胞内のグルコース-6-リン酸の蓄積とピルビン酸への二次シャントが原因である可能性もあり、ピルビン酸はアセチルCoAに変換され、細胞質に運ばれて脂肪酸とコレステロールの合成が行われる。 トリグリセリドが3.4mmol/L(300mg/dL)を超えると、目に見える脂肪血症となり、血漿中の水性画分が減少するため、軽い偽性低ナトリウム血症さえも引き起こすことがある。 GSD Iでは、コレステロールは他の脂質と比較して一般的に軽度しか上昇しない。
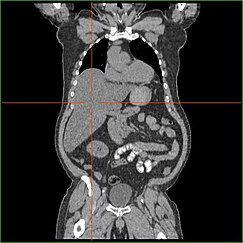
肝臓の糖新生機能が低下すると、臨床的に明らかな肝腫大が起こります。 この過程がないと、体は肝臓からグリコーゲンを遊離させて血糖に変換することができず、肝臓に貯蔵グリコーゲンが蓄積されることになります。 肝臓に蓄積されたグリコーゲンによる肝腫大は、非アルコール性脂肪性肝疾患の一種と考えられています。 GSD Iの患者さんは生涯を通じてある程度の肝腫大を示しますが、重症度は食事性糖質の過剰摂取に関係することが多いようです。 ほとんどの患者は、限られた割合で貯蔵グリコーゲンの遊離を可能にする残存肝機能を保持しているため、肝臓の質量を減少させることが可能である
GSD I患者は、出生時からしばしば肝腫大を呈する。 胎児期には母体のグルコースが胎児に移行して低血糖を防ぐが、グルコースがグリコーゲンとして肝臓に貯蔵されるため、肝腫大が起こる。
GSD type Iの肝腫大は一般に脾臓の交感神経性肥大を伴わずに発生する。 GSD Ib型患者は脾腫を呈することがあるが、これはこの亜型の好中球減少症を治療するためにフィルグラスチムを使用することと関係があり、肝腫大を併発することはない。 肝腫大は生涯を通じてある程度持続し、しばしば腹部が突出し、重症例ではへその下あたりで触知できることがある。 GSD関連非アルコール性脂肪性肝疾患では、通常、肝酵素やビリルビンは正常範囲にとどまり、肝機能は免除されます。
肝腺腫編集
GSD Iの肝腺腫の特異的な病因は、現在研究が進められているものの、いまだ不明である。 少なくとも1つの腺腫を呈する典型的なGSD I患者は成人であるが、病変は14歳の患者にも観察されている。 腺腫は異質な新生物から成り、単独または多発性に発生することがある。 GSD Iにおける肝細胞腺腫から肝細胞癌への転換率の推定値は0%から11%であり、後者はより最近の研究によるものである。
治療基準では、構造的な異常を監視するためにMRIまたはCTスキャンで肝臓を定期的に観察することが指示されている。 肝腺腫は、画像診断において巣状結節性過形成と誤認されることがあるが、このような状態はまれである。 しかしながら、GSD Iの肝腺腫は、限局性結節性過形成によくみられるびまん性マロリーヒアルロン酸沈着を伴うのが特徴である。 経口避妊薬に関連した一般的な肝腺腫とは異なり、GSD I患者における出血はまれである。
GSD Iにおける腺腫の高い有病率の理由は不明であるが、1970年代以降の研究により、血清グルカゴンが潜在的推進要因として関与していることが示唆されている。 血糖値を72~108 mg/dL(4.0~6.0 mmol/L)の正常範囲に保つよう食事療法を行った患者では、腺腫を発症する可能性が低いことが研究で示された。 さらに、血糖が十分にコントロールされている患者では、一貫して肝腺腫のサイズと数が減少しており、腺腫は肝臓における血清インスリンや特に血清グルカゴンなどの肝向性物質の不均衡が原因である可能性が示唆されている<3318><2789>門脈高血圧症編集<5119><1801><7019>このセクションは空です。 追加することによって助けることができます。 (2020年4月)
骨減少症編集
GSD Iの患者さんは、しばしば骨減少症を発症します。 GSDにおける低骨密度の特異的な病因は不明であるが、代謝コントロールの不良と強く関連している。 骨減少症は低血糖が直接の原因である場合もあれば、内分泌や代謝の後遺症が原因である場合もある。 代謝管理の改善は、GSD患者において臨床的に重要な骨減少を予防または回復させることが一貫して示されてきた。 加齢に伴い骨減少が進行する症例では、肋骨の骨密度は椎骨よりも一般的に深刻である。 骨密度Tスコアが-2.5を下回る症例もあり、骨粗鬆症を示唆する。 骨減少症はGSD Iの関連する腎臓の異常、特に濾過腺過多と関連している可能性があるという証拠がいくつかある。 また、この状態はカルシウムの補給に反応するようである。 多くの場合、適切な代謝制御とカルシウムの補給のみで、骨密度は増加し正常範囲に戻り、骨減少症は逆転する。
腎臓への影響編集
腎臓は通常、貯蔵グリコーゲンで10~20%肥大する。 成人のGSD Iでは、糖尿病性腎症に類似した慢性糸球体障害が生じ、腎不全に至ることがある。 GSDⅠ型では様々な腎臓の合併症を呈することがあります。 高乳酸血症に関連した腎尿細管異常は、小児期に乳酸アシドーシスが遷延しやすいためか、早期に認められます。 これは、重炭酸塩とリン酸塩の消耗を伴う尿細管性アシドーシスを含む、腎尿細管再吸収の複数の異常を伴うFanconi症候群として現れることが多いでしょう。 GSD Iにおけるこれらの尿細管異常は、通常、尿中カルシウムによって検出・監視される。 長期にわたるこれらの異常は、高乳酸血症によって引き起こされる尿酸腎症を増悪させる可能性がある。 青年期以降では、糸球体疾患が独立して発症することがあり、最初は尿中eGFRの上昇によって示される糸球体過濾過として現れる。 GSD Iaでは、脾腫は肝臓と脾臓の関係で、どちらかが他方の相対的な大きさに合わせて大きくなったり小さくなったりして、程度が弱くなることが原因である場合がある。
腸管への影響編集
腸管への関与は脂っこい便を伴う軽度の吸収不良を引き起こすことがあるが、通常は治療を必要としない。
感染リスク編集
中性脂肪減少はGSD Iaにはなく、Ibの特徴的な症状である。 GSD Ibにおける好中球減少の微生物学的原因はよく理解されていない。 大まかには、好中球の細胞代謝が損なわれ、好中球のアポトーシスが促進されることから問題が生じると考えられています。 GSDにおける好中球減少症は、好中球の絶対数の減少と好中球の機能低下の両方によって特徴付けられる。 好中球は、細胞内のエネルギー恒常性を維持するために、G6Pase-βまたはG6PTの存在に依存する特定のG6P代謝経路を使用します。 GSD IbのG6PTの欠如は、この経路を制限し、小胞体ストレス、好中球内の酸化ストレスを引き起こし、早期のアポトーシスを誘発する。 顆粒球コロニー刺激因子(G-CSF)は、フィルグラスチムとして販売されており、感染症のリスクを低減することができます。
血小板減少症および血液凝固障害編集
血小板凝集障害は、GSD I患者に見られる慢性低血糖のまれな結果である。 プロトロンビン消費量の減少、異常な凝集反応、出血時間の延長、および血小板接着性の低下によって特徴づけられる血小板機能の低下が研究によって証明されている。 血小板機能障害の重症度は、一般的に臨床状態と相関しており、最も重症なケースは、乳酸アシドーシスや重度の脂質異常症と相関しています。 臨床的に重大な出血、特に鼻出血の原因となることがあります。 さらに、GSD I の患者は、脾臓肥大の結果として血小板減少症を呈することがある。 脾臓肥大の場合、血液が脾臓で濾過されるため、様々な血液学的因子が脾臓の組織に隔離される可能性がある。
発達への影響編集
発達遅延は慢性または再発性の低血糖症の潜在的な副次的影響であるが、少なくとも理論的には予防可能である。 正常な神経細胞や筋肉細胞はグルコース-6-ホスファターゼを発現しないため、GSDⅠの影響を直接受けることはない。 しかし、低血糖を適切に治療しないと、慢性的なインスリンレベルの低下、持続的なアシドーシス、異化ホルモンの慢性的な上昇、カロリー不足(または吸収不良)により、一般に成長不全が引き起こされる。 最も劇的な発育遅延は、しばしば低血糖症の重症エピソード(単に持続的なものではない)の原因である
。