- Echilibrul normal al carbohidraților și menținerea nivelului de glucoză din sângeEdit
- FiziopatologieEdit
- Lactat ridicat și acidoză lacticăEdit
- Urat ridicat și complicațiiEdit
- Hiperlipidemie și efecte plasmaticeEdit
- HepatomegalieEdit
- Adenoame hepaticeEdit
- Hipertensiunea portalăEdit
- OsteopenieEdit
- Efecte renaleEdit
- SplenomegalieEdit
- Efecte intestinaleEdit
- Risc de infecțieEdit
- Trombocitopenie și probleme de coagulare a sângeluiEdit
- Efecte asupra dezvoltăriiEdit
Echilibrul normal al carbohidraților și menținerea nivelului de glucoză din sângeEdit
Glicogenul din ficat și (într-o măsură mai mică) din rinichi servește ca o formă de glucoză stocată, rapid accesibilă, astfel încât nivelul de glucoză din sânge poate fi menținut între mese. Timp de aproximativ 3 ore după o masă care conține carbohidrați, nivelurile ridicate de insulină dirijează celulele hepatice să preia glucoza din sânge, să o transforme în gluco-6-fosfat (G6P) cu ajutorul enzimei glucokinază și să adauge moleculele de G6P la capetele lanțurilor de glicogen (sinteza glicogenului). Excesul de G6P este, de asemenea, deviat în producția de trigliceride și exportat pentru a fi depozitat în țesutul adipos sub formă de grăsime.
Când digestia unei mese este completă, nivelul de insulină scade, iar sistemele enzimatice din celulele hepatice încep să elimine moleculele de glucoză din lanțurile de glicogen sub formă de G6P. Acest proces se numește glicogenoliză. G6P rămâne în interiorul celulei hepatice, cu excepția cazului în care fosfatul este scindat de către glucoză-6-fosfatază. Această reacție de defosforilare produce glucoză liberă și anioni PO
4 liberi. Moleculele de glucoză liberă pot fi transportate din celulele hepatice în sânge pentru a menține o aprovizionare adecvată cu glucoză a creierului și a altor organe ale corpului. Glicogenoliza poate asigura necesarul de glucoză al unui organism adult timp de 12-18 ore.
Când postul continuă mai mult de câteva ore, scăderea nivelului de insulină permite catabolismul proteinelor musculare și al trigliceridelor din țesutul adipos. Produsele acestor procese sunt aminoacizi (în principal alanină), acizi grași liberi și acid lactic. Acizii grași liberi din trigliceride sunt transformați în cetone și în acetil-CoA. Aminoacizii și acidul lactic sunt utilizați pentru a sintetiza G6P nou în celulele hepatice prin procesul de gluconeogeneză. Ultima etapă a gluconeogenezei normale, ca și ultima etapă a glicogenolizei, este desfosforilarea G6P de către glucoza-6-fosfataza în glucoză liberă și PO
Din acest motiv, glucoza-6-fosfataza mediază mediază etapa finală, cheie, în ambele procese principale de producere a glucozei în timpul postului. Efectul este amplificat deoarece nivelurile ridicate de glucoză-6-fosfat care rezultă inhibă etapele cheie anterioare atât în glicogenoliză, cât și în gluconeogeneză.
FiziopatologieEdit
Principalele efecte metabolice ale deficitului de glucoză-6-fosfatază sunt hipoglicemia, acidoza lactică, hipertrigliceridemia și hiperuricemia.

Ipoglicemia din GSD I este denumită „de post”, sau „post-absorbție”, de obicei la aproximativ 4 ore după digestia completă a unei mese. Această incapacitate de a menține niveluri adecvate de glucoză în sânge în timpul postului rezultă din afectarea combinată atât a glicogenolizei, cât și a gluconeogenezei. Hipoglicemia de post este adesea cea mai semnificativă problemă în cazul GSD I și, de obicei, problema care duce la diagnosticare. Hipoglicemia cronică produce adaptări metabolice secundare, inclusiv niveluri cronic scăzute de insulină și niveluri ridicate de glucagon și cortizol.
Acidoza lactică apare din afectarea gluconeogenezei. Acidul lactic este generat atât în ficat, cât și în mușchi și este oxidat de NAD+ în acid piruvic și apoi transformat prin calea gluconeogenică în G6P. Acumularea de G6P inhibă conversia lactatului în piruvat. Nivelul acidului lactic crește în timpul postului, pe măsură ce glicemia scade. La persoanele cu GSD I, este posibil ca acesta să nu scadă în întregime la normal chiar și atunci când sunt restabilite nivelurile normale de glucoză.
Hipertrigliceridemia rezultată din producția amplificată de trigliceride este un alt efect indirect al gluconeogenezei deficitare, amplificat de nivelurile cronic scăzute de insulină. În timpul postului, este afectată conversia normală a trigliceridelor în acizi grași liberi, cetone și, în final, acetil-CoA. Nivelurile de trigliceride în GSD I pot ajunge de câteva ori mai mari decât normalul și servesc ca indice clinic de „control metabolic”.
Hiperuricemia rezultă dintr-o combinație de generare crescută și excreție scăzută de acid uric, care este generat atunci când cantități crescute de G6P sunt metabolizate prin calea pentozei fosfat. Este, de asemenea, un produs secundar al degradării purinelor. Acidul uric concurează cu acidul lactic și alți acizi organici pentru excreția renală în urină. În cazul GSD I, disponibilitatea crescută a G6P pentru calea pentozei fosfat, ratele crescute de catabolism și scăderea excreției urinare din cauza nivelurilor ridicate de acid lactic se combină pentru a produce niveluri de acid uric de câteva ori mai mari decât cele normale. Deși hiperuricemia este asimptomatică timp de ani de zile, leziunile renale și articulare se acumulează treptat.
Lactat ridicat și acidoză lacticăEdit
Niveluri ridicate de acid lactic în sânge sunt observate la toate persoanele cu GSD I, din cauza alterării gluconeogenezei. Nivelurile inițiale variază, în general, între 4 și 10 mol/mL, ceea ce nu va cauza niciun impact clinic. Cu toate acestea, în timpul și după un episod de glicemie scăzută, nivelurile de lactat vor crește brusc pentru a depăși 15 mol/mL, pragul pentru acidoză lactică. Simptomele acidozei lactice includ vărsături și hiperpnee, ambele putând exacerba hipoglicemia în cadrul GSD I. În cazurile de acidoză lactică acută, pacienții au nevoie de îngrijiri de urgență pentru a stabiliza oxigenul din sânge, și a restabili glicemia. Identificarea corectă a acidozei lactice la copiii nediagnosticați reprezintă o provocare, deoarece primele simptome sunt, de obicei, vărsăturile și deshidratarea, ambele imitând infecții ale copilăriei, cum ar fi gastroenterita sau pneumonia. Mai mult, ambele aceste infecții comune pot precipita o hipoglicemie mai severă la copiii nediagnosticați, ceea ce face dificilă diagnosticarea cauzei de bază.
Pe măsură ce lactația crescută persistă, acidul uric, cetoacizii și acizii grași liberi cresc și mai mult diferența anionică. La adulți și copii, concentrațiile ridicate de lactat determină un disconfort semnificativ la nivelul mușchilor. Acest disconfort este o formă amplificată a senzației de arsură pe care un alergător o poate simți în cvadriceps după sprint, care este cauzată de o scurtă acumulare de acid lactic. Controlul adecvat al hipoglicemiei în GSD I elimină în totalitate posibilitatea apariției acidozei lactice.
Urat ridicat și complicațiiEdit
Nivelurile ridicate de acid uric se prezintă adesea ca o consecință a acidului lactic ridicat la pacienții cu GSD I. Atunci când nivelurile de lactat sunt ridicate, acidul lactic transportat prin sânge concurează pentru același mecanism de transport tubular renal ca și uratul, limitând rata cu care uratul poate fi eliminat de rinichi în urină. Dacă este prezent, catabolismul crescut al purinelor este un factor suplimentar care contribuie. Nivelurile de acid uric de 6 până la 12 mg/dl (530 până la 1060 umol/L) sunt frecvente în rândul pacienților cu GSD I, dacă boala nu este tratată corespunzător. La unele persoane afectate, este necesară utilizarea medicamentului alopurinol pentru a reduce nivelul uraților din sânge. Consecințele hiperuricemiei în rândul pacienților cu GSD I includ apariția pietrelor la rinichi și acumularea de cristale de acid uric în articulații, ceea ce duce la afecțiuni renale și, respectiv, gută.
Hiperlipidemie și efecte plasmaticeEdit
Trigliceridele crescute în GSD I rezultă din insulină serică scăzută la pacienții cu hipoglicemie prelungită frecventă. Poate fi cauzată, de asemenea, de acumularea intracelulară de glucoză-6-fosfat cu deviere secundară la piruvat, care este transformat în acetil-CoA, care este transportat în citosol unde are loc sinteza acizilor grași și a colesterolului. Trigliceridele peste intervalul de 3,4 mmol/L (300 mg/dL) pot produce o lipemie vizibilă și chiar o pseudohiponatremie ușoară datorită unei fracții apoase reduse a plasmei sanguine. În GSD I, colesterolul este de obicei doar ușor crescut în comparație cu alte lipide.
HepatomegalieEdit
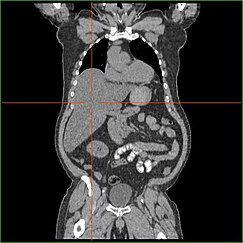
Împiedicarea capacității ficatului de a efectua gluconeogeneza duce la hepatomegalie aparentă clinic. În lipsa acestui proces, organismul este incapabil să elibereze glicogenul din ficat și să îl transforme în glucoză din sânge, ceea ce duce la o acumulare de glicogen stocat în ficat. Hepatomegalia datorată acumulării de glicogen stocat în ficat este considerată o formă de boală hepatică grasă non-alcoolică. Pacienții cu GSD I prezintă un grad de hepatomegalie pe tot parcursul vieții, dar severitatea este adesea legată de consumul de carbohidrați alimentari în exces. Sunt posibile reduceri ale masei ficatului, deoarece majoritatea pacienților păstrează o funcție hepatică reziduală care permite eliberarea glicogenului stocat într-un ritm limitat.
Pacienții cu GSD I prezintă adesea hepatomegalie încă de la naștere. În dezvoltarea fetală, glucoza maternă transferată la făt previne hipoglicemia, dar stocarea glucozei sub formă de glicogen în ficat duce la hepatomegalie. Nu există nicio dovadă că această hepatomegalie prezintă vreun risc pentru dezvoltarea corectă a fătului.
Hepatomegalia în GSD tip I apare în general fără mărirea simpatică a splinei. Pacienții cu GSD Ib pot prezenta splenomegalie, dar aceasta este legată de utilizarea filgrastimului pentru tratarea neutropeniei în acest subtip, nu de hepatomegalia comorbidă. Hepatomegalia va persista într-o oarecare măsură pe tot parcursul vieții, determinând adesea proeminența abdomenului, iar în cazurile severe poate fi palpabilă la sau sub buric. În boala hepatică grasă non-alcoolică legată de GSD, funcția hepatică este de obicei cruțată, enzimele hepatice și bilirubina rămânând în limitele normale. Cu toate acestea, funcția hepatică poate fi afectată de alte complicații hepatice la vârsta adultă, inclusiv dezvoltarea adenoamelor hepatice.
Adenoame hepaticeEdit
Etiologia specifică a adenoamelor hepatice la GSD I rămâne necunoscută, în ciuda cercetărilor în curs. Pacientul tipic GSD I care prezintă cel puțin un adenom este un adult, deși au fost observate leziuni la pacienți cu vârsta de doar 14 ani. Adenoamele, compuse din neoplazii eterogene, pot apărea individual sau în multiple. Estimările privind rata de conversie a unui adenom hepatocelular în carcinom hepatocelular în GSD I variază între 0% și 11%, ultima cifră reprezentând cercetări mai recente. Un motiv pentru estimarea în creștere este populația tot mai mare de pacienți cu GSD I care supraviețuiesc până la vârsta adultă, când se dezvoltă majoritatea adenoamelor.
Standardele de tratament impun observarea periodică a ficatului prin RMN sau CT pentru a monitoriza anomaliile structurale. Adenoamele hepatice pot fi identificate greșit ca hiperplazie nodulară focală în imagistica de diagnostic, deși această afecțiune este rară. Cu toate acestea, adenoamele hepatice în GSD I implică în mod unic depunerea hialină Mallory difuză, care este altfel frecvent observată în hiperplazia nodulară focală. Spre deosebire de adenoamele hepatice comune legate de contracepția orală, hemoragia la pacienții cu GSD I este rară.
În timp ce motivul prevalenței ridicate a adenoamelor în GSD I este neclar, cercetările efectuate începând cu anii 1970 au implicat glucagonul seric ca fiind un potențial factor determinant. În cadrul studiilor, pacienții care au fost supuși unui regim alimentar pentru a menține glicemia într-un interval normal cuprins între 72 și 108 mg/dL (4,0 și 6,0 mmol/L) au prezentat o probabilitate redusă de a dezvolta adenoame. Mai mult, pacienții cu glicemia bine controlată au înregistrat în mod constant o reducere a dimensiunii și a numărului de adenoame hepatice, sugerând că adenoamele pot fi cauzate de dezechilibre ale agenților hepatotropi, cum ar fi insulina serică și mai ales glucagonul seric din ficat.
Hipertensiunea portalăEdit
OsteopenieEdit
Pacienții cu GSD I vor dezvolta adesea osteopenie. Etiologia specifică a densității minerale osoase scăzute în GSD nu este cunoscută, deși este puternic asociată cu un control metabolic deficitar. Osteopenia poate fi cauzată direct de hipoglicemie, sau de sechelele endocrine și metabolice rezultate. S-a demonstrat în mod constant că îmbunătățirile controlului metabolic previn sau inversează osteopenia relevantă din punct de vedere clinic la pacienții cu GSD I. În cazurile în care osteopenia progresează odată cu vârsta, densitatea minerală osoasă la nivelul coastelor este de obicei mai severă decât la nivelul vertebrelor. În unele cazuri, scorul T al densității minerale osoase va scădea sub -2,5, indicând osteoporoză. Există unele dovezi că osteopenia poate fi legată de anomalii renale asociate la GSD I, în special de hiperfiltrarea glomerulară. Afecțiunea pare să răspundă, de asemenea, la suplimentarea cu calciu. În multe cazuri, densitatea minerală osoasă poate crește și reveni la intervalul normal, dat fiind un control metabolic adecvat și suplimentarea cu calciu singur, inversând osteopenia.
Efecte renaleEdit
Regii sunt de obicei măriți cu 10 până la 20% cu glicogenul stocat. La adulții cu GSD I, afectarea glomerulară cronică similară nefropatiei diabetice poate duce la insuficiență renală. GSD I se poate prezenta cu diverse complicații renale. Anomaliile tubulare renale legate de hiperlactatemie sunt observate devreme în viață, probabil pentru că acidoza lactică prelungită este mai probabil să apară în copilărie. Aceasta se va prezenta adesea sub forma sindromului Fanconi cu dereglări multiple ale reabsorbției tubulare renale, inclusiv acidoză tubulară cu pierdere de bicarbonat și fosfat. Aceste anomalii tubulare în GSD I sunt de obicei detectate și monitorizate prin calciu urinar. Pe termen lung, aceste dereglări pot exacerba nefropatia cu acid uric, altfel determinată de hiperlactatemie. În adolescență și ulterior, boala glomerulară se poate dezvolta în mod independent, prezentându-se inițial ca hiperfiltrare glomerulară indicată de un eGFR urinar ridicat.
SplenomegalieEdit
Lărgirea splinei (splenomegalie) este frecventă în GSD I și are două cauze primare. La GSD Ia, splenomegalia poate fi cauzată de o relație între ficat și splină care face ca oricare dintre ele să crească sau să se micșoreze pentru a se potrivi cu dimensiunea relativă a celeilalte, la un grad diminuat. În GSD Ib, este un efect secundar al utilizării filgrastimului pentru a trata neutropenia.
Efecte intestinaleEdit
Aplicarea intestinală poate cauza malabsorbție ușoară cu scaune grase (steatorree), dar de obicei nu necesită tratament.
Risc de infecțieEdit
Neutropenia este o caracteristică distinctivă a GSD Ib, absentă în GSD Ia. Cauza microbiologică a neutropeniei în GSD Ib nu este bine înțeleasă. În linii mari, problema apare din cauza metabolismului celular compromis la nivelul neutrofilelor, ceea ce duce la apoptoza accelerată a neutrofilelor. Neutropenia în GSD se caracterizează atât prin scăderea numărului absolut de neutrofile, cât și prin diminuarea funcției neutrofilelor. Neutrofilele utilizează o cale metabolică specifică a G6P care se bazează pe prezența G6Pazei-β sau G6PT pentru a menține homeostazia energetică în interiorul celulei. Absența G6PT în GSD Ib limitează această cale, ceea ce duce la stresul reticulului endoplasmatic, stresul oxidativ în cadrul neutrofilelor, declanșând apoptoza prematură. Factorul de stimulare a coloniilor de granulocite (G-CSF), disponibil sub formă de filgrastim, poate reduce riscul de infecție. În unele cazuri, G-CSF formulat sub formă de pegfilgrastim, comercializat sub denumirea comercială Neulasta, poate fi utilizat ca o alternativă cu acțiune lentă, necesitând doze mai puțin frecvente.
Trombocitopenie și probleme de coagulare a sângeluiEdit
Agregarea trombocitelor este o consecință mai puțin frecventă a hipoglicemiei cronice, observată la pacienții cu GSD I. Cercetările au demonstrat scăderea funcției plachetare, caracterizată prin scăderea consumului de protrombină, reacții anormale de agregare, timp de sângerare prelungit și aderență plachetară scăzută. Severitatea disfuncției plachetare se corelează de obicei cu starea clinică, cele mai grave cazuri fiind corelate cu acidoza lactică și lipidemia severă. Poate provoca sângerări semnificative din punct de vedere clinic, în special epistaxis. În plus, pacienții cu GSD I pot prezenta trombocitopenie ca o consecință a splenomegaliei. În cadrul splenomegaliei, diverși factori hematologici pot fi sechestrați în țesuturile splinei, deoarece sângele este filtrat prin acest organ. Acest lucru poate diminua nivelurile de trombocite disponibile în fluxul sanguin, ducând la trombocitopenie.
Efecte asupra dezvoltăriiEdit
Retârzierea dezvoltării este un potențial efect secundar al hipoglicemiei cronice sau recurente, dar este cel puțin teoretic prevenibilă. Celulele neuronale și musculare normale nu exprimă glucoza-6-fosfataza și, prin urmare, nu sunt afectate direct de GSD I. Cu toate acestea, fără un tratament adecvat al hipoglicemiei, eșecul de creștere rezultă în mod obișnuit din nivelurile cronic scăzute de insulină, acidoza persistentă, creșterea cronică a hormonilor catabolici și insuficiența calorică (sau malabsorbția). Cele mai dramatice întârzieri de dezvoltare sunt adesea cauza unor episoade severe (nu doar persistente) de hipoglicemie.
.