- Normalna równowaga węglowodanowa i utrzymanie poziomu glukozy we krwiEdit
- PatofizjologiaEdit
- Podwyższony mleczan i kwasica mleczanowaEdit
- Podwyższony poziom moczanów i powikłaniaEdit
- Hiperlipidemia i wpływ na osocze krwiEdit
- HepatomegaliaEdit
- Gruczolaki wątrobyEdit
- Nadciśnienie wrotneEdit
- OsteopeniaEdit
- Wpływ na nerkiEdit
- SplenomegaliaEdit
- Wpływ na jelitaEdit
- Ryzyko zakażeniaEdit
- Małopłytkowość i zaburzenia krzepnięcia krwiEdit
- Skutki rozwojoweEdit
Normalna równowaga węglowodanowa i utrzymanie poziomu glukozy we krwiEdit
Glikogen w wątrobie i (w mniejszym stopniu) nerkach służy jako forma przechowywanej, szybko dostępnej glukozy, dzięki czemu poziom glukozy we krwi może być utrzymywany między posiłkami. Przez około 3 godziny po posiłku zawierającym węglowodany, wysoki poziom insuliny kieruje komórki wątroby do pobierania glukozy z krwi, przekształcania jej w glukozo-6-fosforan (G6P) za pomocą enzymu glukokinazy i dodawania cząsteczek G6P do końców łańcuchów glikogenu (synteza glikogenu). Nadmiar G6P jest również kierowany do produkcji triglicerydów i eksportowany do przechowywania w tkance tłuszczowej jako fat.
Gdy trawienie posiłku jest zakończone, poziom insuliny spada, a systemy enzymatyczne w komórkach wątroby zaczynają usuwać cząsteczki glukozy z łańcuchów glikogenu w postaci G6P. Proces ten określany jest mianem glikogenolizy. G6P pozostaje w komórce wątroby, dopóki fosforan nie zostanie rozłożony przez glukozo-6-fosfatazę. W wyniku reakcji deposforylacji powstaje wolna glukoza i wolne aniony PO
4. Wolne cząsteczki glukozy mogą być transportowane z komórek wątroby do krwi w celu utrzymania odpowiedniego zaopatrzenia w glukozę mózgu i innych narządów organizmu. Glikogenoliza może zaspokoić zapotrzebowanie na glukozę dorosłego organizmu przez 12-18 godzin.
Gdy post trwa dłużej niż kilka godzin, spadający poziom insuliny pozwala na katabolizm białek mięśniowych i trójglicerydów z tkanki tłuszczowej. Produktami tych procesów są aminokwasy (głównie alanina), wolne kwasy tłuszczowe i kwas mlekowy. Wolne kwasy tłuszczowe z trójglicerydów są przekształcane do ketonów, a także do acetylo-CoA. Aminokwasy i kwas mlekowy są wykorzystywane do syntezy nowego G6P w komórkach wątroby w procesie glukoneogenezy. Ostatnim etapem normalnej glukoneogenezy, podobnie jak ostatnim etapem glikogenolizy, jest dephosphorylation of G6P by glucose-6-phosphatase to free glucose and PO
Thus glucose-6-phosphatase mediates the final, key, step in both of the two main processes of glucose production during fasting. Efekt ten jest wzmocniony, ponieważ wynikające z tego wysokie poziomy glukozo-6-fosforanu hamują wcześniejsze kluczowe etapy zarówno glikogenolizy, jak i glukoneogenezy.
PatofizjologiaEdit
Głównymi skutkami metabolicznymi niedoboru glukozo-6-fosfatazy są hipoglikemia, kwasica mleczanowa, hipertriglicerydemia i hiperurykemia.

Hipoglikemia w GSD I jest określana jako „na czczo”, lub „poabsorpcyjna”, zwykle około 4 godzin po całkowitym strawieniu posiłku. Niezdolność do utrzymania odpowiedniego poziomu glukozy we krwi podczas postu wynika z łącznego upośledzenia zarówno glikogenolizy, jak i glukoneogenezy. Hipoglikemia na czczo jest często najistotniejszym problemem w GSD I, i zazwyczaj jest to problem, który prowadzi do postawienia diagnozy. Przewlekła hipoglikemia powoduje wtórne adaptacje metaboliczne, w tym chronicznie niski poziom insuliny oraz wysoki poziom glukagonu i kortyzolu.
Kwasica mleczanowa powstaje w wyniku upośledzenia glukoneogenezy. Kwas mlekowy jest generowany zarówno w wątrobie jak i mięśniach i jest utleniany przez NAD+ do kwasu pirogronowego, a następnie przekształcany przez szlak glukoneogenetyczny do G6P. Akumulacja G6P hamuje konwersję mleczanu do pirogronianu. Poziom kwasu mlekowego wzrasta na czczo, gdy spada poziom glukozy. U osób z GSD I może on nie spaść całkowicie do normy, nawet gdy przywrócony zostanie prawidłowy poziom glukozy.
Hypertriglicerydemia wynikająca ze zwiększonej produkcji triglicerydów jest kolejnym pośrednim skutkiem upośledzonej glukoneogenezy, potęgowanej przez chronicznie niski poziom insuliny. Podczas postu normalna konwersja triglicerydów do wolnych kwasów tłuszczowych, ketonów i ostatecznie acetylo-CoA jest zaburzona. Poziom triglicerydów w GSD I może osiągnąć kilka razy normalny i służyć jako kliniczny wskaźnik „kontroli metabolicznej”.
Nadciśnienie wynika z połączenia zwiększonego wytwarzania i zmniejszonego wydalania kwasu moczowego, który jest wytwarzany, gdy zwiększone ilości G6P są metabolizowane przez szlak fosforanu pentozy. Jest on również produktem ubocznym degradacji puryn. Kwas moczowy konkuruje z kwasem mlekowym i innymi kwasami organicznymi o wydalanie z moczem. W GSD I zwiększona dostępność G6P dla szlaku fosforanu pentozy, zwiększone tempo katabolizmu i zmniejszone wydalanie z moczem z powodu wysokiego poziomu kwasu mlekowego powodują, że stężenie kwasu moczowego jest kilkakrotnie wyższe od normalnego. Chociaż hiperurykemia jest bezobjawowa przez lata, uszkodzenia nerek i stawów stopniowo narastają.
Podwyższony mleczan i kwasica mleczanowaEdit
Wysoki poziom kwasu mlekowego we krwi obserwuje się u wszystkich osób z GSD I, z powodu upośledzonej glukoneogenezy. Poziomy wyjściowe wynoszą zazwyczaj od 4 do 10 mol/mL, co nie powoduje żadnych skutków klinicznych. Jednakże, podczas i po epizodzie niskiego poziomu cukru we krwi, poziom mleczanów gwałtownie wzrasta, przekraczając 15 mol/mL, co stanowi próg dla kwasicy mleczanowej. Objawami kwasicy mleczanowej są wymioty i hiperpnea, które mogą nasilać hipoglikemię w przebiegu GSD I. W przypadku ostrej kwasicy mleczanowej pacjenci wymagają pomocy doraźnej, aby ustabilizować poziom tlenu we krwi i przywrócić poziom glukozy we krwi. Właściwe rozpoznanie kwasicy mleczanowej u niezdiagnozowanych dzieci stanowi wyzwanie, ponieważ pierwszymi objawami są zwykle wymioty i odwodnienie, które imitują infekcje dziecięce, takie jak zapalenie żołądka i jelit lub zapalenie płuc. Co więcej, obie te częste infekcje mogą poprzedzać cięższą hipoglikemię u niezdiagnozowanych dzieci, co utrudnia rozpoznanie przyczyny.
Jak podwyższony mleczan utrzymuje się, kwas moczowy, ketokwasy i wolne kwasy tłuszczowe dodatkowo zwiększają lukę anionową. U dorosłych i dzieci wysokie stężenie mleczanów powoduje znaczny dyskomfort w mięśniach. Dyskomfort ten jest wzmocnioną formą uczucia pieczenia, jakie biegacz może odczuwać w mięśniu czworogłowym po sprincie, które jest spowodowane krótkotrwałym nagromadzeniem się kwasu mlekowego. Prawidłowa kontrola hipoglikemii w GSD I całkowicie eliminuje możliwość wystąpienia kwasicy mleczanowej.
Podwyższony poziom moczanów i powikłaniaEdit
Wysoki poziom kwasu moczowego często występuje jako konsekwencja podwyższonego poziomu kwasu mlekowego u pacjentów z GSD I. Kiedy poziom mleczanów jest podwyższony, kwas mlekowy we krwi konkuruje o ten sam mechanizm transportu kanalikowego w nerkach, co moczany, ograniczając szybkość, z jaką moczany mogą być usuwane przez nerki do moczu. Jeśli jest obecny, dodatkowym czynnikiem przyczyniającym się jest zwiększony katabolizm puryn. Poziom kwasu moczowego wynoszący od 6 do 12 mg/dl (530 do 1060 umol/l) jest powszechny u pacjentów z GSD I, jeśli choroba nie jest odpowiednio leczona. U niektórych chorych konieczne jest stosowanie leku allopurinol w celu obniżenia poziomu moczanów we krwi. Konsekwencje hiperurykemii u pacjentów z GSD I obejmują rozwój kamieni nerkowych i odkładanie się kryształów kwasu moczowego w stawach, co prowadzi odpowiednio do choroby nerek i podagry.
Hiperlipidemia i wpływ na osocze krwiEdit
Podwyższone stężenie trójglicerydów w GSD I wynika z niskiego stężenia insuliny w surowicy krwi u pacjentów z częstą, przedłużającą się hipoglikemią. Może być również spowodowane wewnątrzkomórkowym gromadzeniem się glukozo-6-fosforanu z wtórnym przejściem do pirogronianu, który jest przekształcany w acetylo-CoA, który jest transportowany do cytozolu, gdzie zachodzi synteza kwasów tłuszczowych i cholesterolu. Trójglicerydy powyżej 3,4 mmol/l (300 mg/dl) mogą powodować widoczną lipemię, a nawet łagodną pseudohyponatremię z powodu zmniejszonej frakcji wodnej osocza krwi. W GSD I cholesterol jest zwykle tylko łagodnie podwyższony w porównaniu z innymi lipidami.
HepatomegaliaEdit
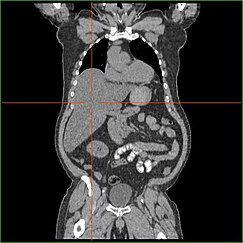
Upośledzenie zdolności wątroby do przeprowadzania glukoneogenezy prowadzi do klinicznie widocznej hepatomegalii. Bez tego procesu organizm nie jest w stanie uwolnić glikogenu z wątroby i przekształcić go w glukozę we krwi, co prowadzi do nagromadzenia zmagazynowanego glikogenu w wątrobie. Hepatomegalia wynikająca z nagromadzenia zmagazynowanego glikogenu w wątrobie jest uważana za formę niealkoholowej choroby stłuszczeniowej wątroby. U pacjentów z GSD I hepatomegalia występuje w pewnym stopniu przez całe życie, ale jej nasilenie jest często związane ze spożywaniem nadmiaru węglowodanów w diecie. Zmniejszenie masy wątroby jest możliwe, ponieważ większość pacjentów zachowuje resztkową funkcję wątroby, która pozwala na uwalnianie zmagazynowanego glikogenu w ograniczonym tempie.
Pacjenci z GSD I często mają hepatomegalię od urodzenia. W rozwoju płodowym, matczyna glukoza przekazywana płodowi zapobiega hipoglikemii, ale magazynowanie glukozy w postaci glikogenu w wątrobie prowadzi do hepatomegalii. Nie ma dowodów na to, że hepatomegalia ta stanowi jakiekolwiek zagrożenie dla prawidłowego rozwoju płodu.
Hepatomegalia w GSD typu I występuje na ogół bez współczulnego powiększenia śledziony. U pacjentów z GSD Ib może wystąpić powiększenie śledziony, ale jest to związane ze stosowaniem filgrastimu w leczeniu neutropenii w tym podtypie, a nie z towarzyszącą hepatomegalią. Hepatomegalia utrzymuje się do pewnego stopnia przez całe życie, często powodując uwypuklenie brzucha, a w ciężkich przypadkach może być wyczuwalna na wysokości lub poniżej pępka. W przypadku niealkoholowej stłuszczeniowej choroby wątroby związanej z GSD, czynność wątroby jest zazwyczaj prawidłowa, enzymy wątrobowe i bilirubina pozostają w granicach normy. Jednakże w wieku dorosłym na czynność wątroby mogą wpływać inne powikłania wątrobowe, w tym rozwój gruczolaków wątroby.
Gruczolaki wątrobyEdit
Szczególna etiologia gruczolaków wątroby w GSD I pozostaje nieznana, pomimo trwających badań. Typowy pacjent z GSD I prezentujący co najmniej jednego gruczolaka jest dorosły, chociaż zmiany obserwowano u pacjentów nawet w wieku 14 lat. Gruczolaki, składające się z heterogennych nowotworów, mogą występować pojedynczo lub wielokrotnie. Szacunki dotyczące częstości konwersji gruczolaka wątrobowokomórkowego w raka wątrobowokomórkowego w GSD I wahają się od 0% do 11%, przy czym ta ostatnia wartość jest wynikiem nowszych badań. Jedną z przyczyn rosnących szacunków jest rosnąca populacja pacjentów z GSD I, którzy przeżywają do wieku dorosłego, kiedy to rozwija się większość gruczolaków.
Standardy leczenia nakazują regularną obserwację wątroby za pomocą rezonansu magnetycznego lub tomografii komputerowej w celu monitorowania nieprawidłowości strukturalnych. Gruczolaki wątroby mogą być błędnie zidentyfikowane jako ogniskowa hiperplazja guzkowa w diagnostyce obrazowej, chociaż ten stan jest rzadki. Jednakże, gruczolaki wątroby w GSD I wyjątkowo charakteryzują się rozproszonym odkładaniem hialiny Mallory’ego, co w innych przypadkach jest powszechnie obserwowane w ogniskowej hiperplazji guzkowej. W przeciwieństwie do częstych gruczolaków wątroby związanych z doustną antykoncepcją, krwotok u pacjentów z GSD I jest rzadki.
Przyczyna wysokiej częstości występowania gruczolaków w GSD I jest niejasna, badania prowadzone od lat 70-tych XX wieku wskazują na glukagon w surowicy jako potencjalny czynnik sprawczy. W badaniach, pacjenci, które zostały wprowadzone na reżim diety, aby utrzymać poziom cukru we krwi w normalnym zakresie obejmującym 72 do 108 mg/dl (4.0 do 6.0 mmol/L) wykazały zmniejszone prawdopodobieństwo rozwoju gruczolaków. Ponadto u pacjentów z dobrze kontrolowanym stężeniem glukozy we krwi konsekwentnie obserwowano zmniejszenie wielkości i liczby gruczolaków wątroby, co sugeruje, że gruczolaki mogą być spowodowane zaburzeniami równowagi czynników hepatotropowych, takich jak insulina w surowicy, a zwłaszcza glukagon w wątrobie.
Nadciśnienie wrotneEdit
OsteopeniaEdit
Pacjenci z GSD I często będą rozwijać osteopenię. Specyficzna etiologia niskiej gęstości mineralnej kości w GSD nie jest znana, choć jest ona silnie związana ze słabą kontrolą metaboliczną. Osteopenia może być bezpośrednio spowodowana hipoglikemią lub wynikać z następstw endokrynologicznych i metabolicznych. Wykazano, że poprawa kontroli metabolicznej konsekwentnie zapobiega lub odwraca klinicznie istotną osteopenię u pacjentów z GSD I. W przypadkach, w których osteopenia postępuje wraz z wiekiem, gęstość mineralna kości w żebrach jest zwykle bardziej nasilona niż w kręgach. W niektórych przypadkach wskaźnik T-score gęstości mineralnej kości spada poniżej -2,5, co wskazuje na osteoporozę. Istnieją pewne dowody na to, że osteopenia może być związana z nieprawidłowościami nerek w GSD I, szczególnie z hiperfiltracją kłębuszkową. Stan ten wydaje się również reagować na suplementację wapnia. W wielu przypadkach gęstość mineralna kości może wzrosnąć i powrócić do normalnego zakresu przy odpowiedniej kontroli metabolicznej i samej suplementacji wapnia, odwracając osteopenię.
Wpływ na nerkiEdit
Nerki są zwykle 10 do 20% powiększone o zmagazynowany glikogen. U dorosłych z GSD I, przewlekłe uszkodzenie kłębuszków nerkowych podobne do nefropatii cukrzycowej może prowadzić do niewydolności nerek. GSD I może przebiegać z różnymi powikłaniami ze strony nerek. Zaburzenia kanalików nerkowych związane z hiperlaktatemią obserwuje się we wczesnym okresie życia, prawdopodobnie dlatego, że przedłużająca się kwasica mleczanowa jest bardziej prawdopodobna w dzieciństwie. Często objawia się to jako zespół Fanconiego z licznymi zaburzeniami wchłaniania zwrotnego w kanalikach nerkowych, w tym kwasicą kanalikową z utratą wodorowęglanów i fosforanów. Te nieprawidłowości kanalikowe w GSD I są zwykle wykrywane i monitorowane na podstawie stężenia wapnia w moczu. Długotrwałe utrzymywanie się tych zaburzeń może nasilać nefropatię kwasowo-moczową, która w innych przypadkach jest spowodowana hiperlaktatemią. W okresie dojrzewania i później, choroba kłębuszków nerkowych może rozwijać się niezależnie, początkowo prezentując się jako hiperfiltracja kłębuszkowa, na którą wskazuje podwyższony eGFR w moczu.
SplenomegaliaEdit
Powiększenie śledziony (splenomegalia) jest powszechne w GSD I i ma dwie podstawowe przyczyny. W GSD Ia, splenomegalia może być spowodowana relacją pomiędzy wątrobą i śledzioną, która powoduje, że jedna z nich rośnie lub kurczy się, aby dopasować się do względnej wielkości drugiej, w mniejszym stopniu. W GSD Ib jest to efekt uboczny stosowania filgrastimu w leczeniu neutropenii.
Wpływ na jelitaEdit
Zajęcie jelit może powodować łagodne zaburzenia wchłaniania z tłustymi stolcami (steatorrhea), ale zazwyczaj nie wymaga leczenia.
Ryzyko zakażeniaEdit
Neutropenia jest cechą wyróżniającą GSD Ib, nieobecną w GSD Ia. Mikrobiologiczna przyczyna neutropenii w GSD Ib nie jest dobrze poznana. Najogólniej problem wynika z upośledzenia metabolizmu komórkowego neutrofilów, co skutkuje przyspieszoną apoptozą neutrofilów. Neutropenia w GSD charakteryzuje się zarówno spadkiem bezwzględnej liczby neutrofilów, jak i osłabieniem ich funkcji. Neutrofile wykorzystują specyficzny szlak metaboliczny G6P, który opiera się na obecności G6Pase-β lub G6PT w celu utrzymania homeostazy energetycznej wewnątrz komórki. Brak G6PT w GSD Ib ogranicza ten szlak, prowadząc do stresu retikulum endoplazmatycznego, stresu oksydacyjnego w obrębie neutrofili, wyzwalając przedwczesną apoptozę. Czynnik stymulujący tworzenie kolonii granulocytów (G-CSF), dostępny jako filgrastim, może zmniejszyć ryzyko zakażenia. W niektórych przypadkach G-CSF w postaci pegfilgrastymu, sprzedawanego pod nazwą handlową Neulasta, może być stosowany jako wolno działająca alternatywa, wymagająca rzadszego dawkowania.
Małopłytkowość i zaburzenia krzepnięcia krwiEdit
Zaburzona agregacja płytek krwi jest rzadką konsekwencją przewlekłej hipoglikemii, obserwowaną u pacjentów z GSD I. Badania wykazały obniżoną funkcję płytek krwi, charakteryzującą się zmniejszonym zużyciem protrombiny, nieprawidłowymi reakcjami agregacji, wydłużonym czasem krwawienia i niską adhezyjnością płytek. Nasilenie zaburzeń funkcji płytek zwykle koreluje ze stanem klinicznym, przy czym najcięższe przypadki korelują z kwasicą mleczanową i ciężką lipidemią. Może to być przyczyną istotnych klinicznie krwawień, zwłaszcza krwawień z nosa. Dodatkowo u pacjentów z GSD I może wystąpić małopłytkowość jako konsekwencja splenomegalii. W warunkach splenomegalii różne czynniki hematologiczne mogą być sekwestrowane w tkankach śledziony, ponieważ krew jest filtrowana przez ten narząd. Może to zmniejszać poziom płytek krwi dostępnych w krwiobiegu, prowadząc do małopłytkowości.
Skutki rozwojoweEdit
Opóźnienie rozwojowe jest potencjalnym skutkiem wtórnym przewlekłej lub nawracającej hipoglikemii, ale przynajmniej teoretycznie można mu zapobiec. Normalne komórki neuronalne i mięśniowe nie wykazują ekspresji glukozo-6-fosfatazy, a zatem nie są bezpośrednio dotknięte GSD I. Jednakże, bez odpowiedniego leczenia hipoglikemii, niepowodzenie wzrostu powszechnie wynika z chronicznie niskiego poziomu insuliny, utrzymującej się kwasicy, chronicznego podwyższenia poziomu hormonów katabolicznych i niedoboru kalorii (lub złego wchłaniania). Najbardziej dramatyczne opóźnienia rozwojowe są często przyczyną ciężkich (nie tylko uporczywych) epizodów hipoglikemii.
.