- Equilíbrio normal de carboidratos e manutenção dos níveis de glicose no sangueEditar
- FisiopatologiaEditar
- Lactato elevado e acidose lácticaEdito
- Urato elevado e complicaçõesEditar
- Hiperlipidemia e efeitos plasmáticosEditar
- HepatomegalyEdit
- Adenomas hepáticosEditar
- Hipertensão PortalEditar
- OsteopeniaEdit
- Efeitos renaisEditar
- SplenomegalyEdit
- Efeitos intestinaisEdit
- Risco de infecçãoEdit
- Trombocitopenia e problemas de coagulação sanguíneaEditar
- Efeitos no desenvolvimentoEditar
Equilíbrio normal de carboidratos e manutenção dos níveis de glicose no sangueEditar
Glicogênio no fígado e (em menor grau) nos rins serve como uma forma de armazenamento, glicose de acesso rápido, para que o nível de glicose no sangue possa ser mantido entre as refeições. Durante cerca de 3 horas após uma refeição contendo carboidratos, altos níveis de insulina direcionam as células do fígado para retirar a glicose do sangue, para convertê-la em glucose-6-fosfato (G6P) com a enzima glucokinase, e para adicionar as moléculas de G6P às extremidades das cadeias de glicogênio (síntese de glicogênio). O excesso de G6P também é desviado para a produção de triglicerídeos e exportado para armazenamento em tecido adiposo como gordura.
Quando a digestão de uma refeição está completa, os níveis de insulina caem, e os sistemas enzimáticos nas células hepáticas começam a remover as moléculas de glicose dos fios de glicogênio na forma de G6P. Este processo é denominado glicogenólise. O G6P permanece dentro da célula hepática a menos que o fosfato seja clivado pela glucose-6-fosfatase. Esta reação de desfosforilação produz glicose livre e ânions PO
4 livres. As moléculas livres de glucose podem ser transportadas para fora das células hepáticas para o sangue para manter um suprimento adequado de glucose para o cérebro e outros órgãos do corpo. A glicólise pode suprir as necessidades de glicose de um corpo adulto por 12-18 horas.
Quando o jejum continua por mais de algumas horas, a queda dos níveis de insulina permite o catabolismo da proteína muscular e triglicérides do tecido adiposo. Os produtos destes processos são aminoácidos (principalmente alanina), ácidos gordos livres e ácido láctico. Os ácidos gordos livres dos triglicéridos são convertidos em cetonas, e em acetil-CoA. Aminoácidos e ácido láctico são usados para sintetizar novo G6P em células hepáticas através do processo de gluconeogénese. A última etapa da gluconeogênese normal, como a última etapa da glicogênese, é a desfosforilação do G6P pela glucose-6-fosfatase para liberar glicose e PO
Thus glucose-6-fosfatase medeia a etapa final, chave, em ambos os dois principais processos de produção de glicose durante o jejum. O efeito é amplificado porque os altos níveis de glucose-6-fosfato resultantes inibem os passos-chave anteriores tanto na glicogênese quanto na gluconeogênese.
FisiopatologiaEditar
Os principais efeitos metabólicos da deficiência de glucose-6-fosfatase são hipoglicemia, acidose láctica, hipertrigliceridemia e hiperuricemia.

A hipoglicemia de GSD I é denominada “jejum”, ou “pós-absorvente”, geralmente cerca de 4 horas após a digestão completa de uma refeição. Esta incapacidade de manter níveis adequados de glicemia durante o jejum resulta do comprometimento combinado de glicogênese e gluconeogênese. A hipoglicemia em jejum é frequentemente o problema mais significativo no GSD I, e tipicamente o problema que leva ao diagnóstico. A hipoglicemia crônica produz adaptações metabólicas secundárias, incluindo níveis cronicamente baixos de insulina e altos níveis de glucagon e cortisol.
A acidose láctica surge do comprometimento da gluconeogênese. O ácido láctico é gerado tanto no fígado como no músculo e é oxidado por NAD+ para ácido pirúvico e depois convertido através da via gluconeogénica para G6P. A acumulação de G6P inibe a conversão do ácido láctico em piruvato. O nível de ácido láctico sobe durante o jejum, à medida que a glicose cai. Em pessoas com GSD I, ele pode não cair totalmente ao normal mesmo quando os níveis normais de glicose são restaurados.
Hipertrigliceridemia resultante da produção amplificada de triglicerídeos é outro efeito indireto da gluconeogênese comprometida, amplificada por níveis cronicamente baixos de insulina. Durante o jejum, a conversão normal dos triglicerídeos em ácidos graxos livres, cetonas e, finalmente, acetil-CoA é prejudicada. Os níveis de triglicerídeos no GSD I podem atingir várias vezes o normal e servir como índice clínico de “controle metabólico”.
Hiperuricemia resulta de uma combinação de aumento de geração e diminuição da excreção de ácido úrico, que é gerado quando quantidades aumentadas de G6P são metabolizadas através da via de fosfato pentose. É também um subproduto da degradação da purina. O ácido úrico compete com o ácido láctico e outros ácidos orgânicos para a excreção renal na urina. No GSD I aumentou a disponibilidade do G6P para o caminho do fosfato pentose, aumentou as taxas de catabolismo e diminuiu a excreção urinária devido aos altos níveis de ácido láctico, todos se combinam para produzir níveis de ácido úrico várias vezes normais. Embora a hiperuricemia seja assintomática durante anos, os danos renais e articulares aumentam gradualmente.
Lactato elevado e acidose lácticaEdito
Níveis elevados de ácido láctico no sangue são observados em todas as pessoas com GSD I, devido à gluconeogénese deficiente. As elevações de base geralmente variam de 4 a 10 mol/mL, o que não causará qualquer impacto clínico. No entanto, durante e após um episódio de glicemia baixa, os níveis de ácido láctico subirão abruptamente para exceder 15 mol/mL, o limiar para a acidose láctica. Os sintomas de acidose láctica incluem vómitos e hiperpneia, o que pode exacerbar a hipoglicemia no estabelecimento do GSD I. Em casos de acidose láctica aguda, os doentes necessitam de cuidados de emergência para estabilizar o oxigénio no sangue, e restaurar a glicemia. A identificação adequada da acidose láctica em crianças não diagnosticadas representa um desafio, uma vez que os primeiros sintomas são tipicamente vómitos e desidratação, ambos imitando infecções infantis como a gastroenterite ou a pneumonia. Além disso, ambas as infecções comuns podem precipitar hipoglicemia mais grave em crianças não diagnosticadas, dificultando o diagnóstico da causa subjacente.
As elevated lactate persists, uric acid, ketoacids, and free fatty acids increase further the anion gap. Em adultos e crianças, as altas concentrações de lactato causam desconforto significativo nos músculos. Este desconforto é uma forma ampliada da sensação de ardor que um corredor pode sentir no quadríceps após o sprint, que é causado por um breve acúmulo de ácido láctico. O controle adequado da hipoglicemia em GSD I elimina completamente a possibilidade de acidose láctica.
Urato elevado e complicaçõesEditar
Níveis elevados de ácido úrico frequentemente presentes como consequência de ácido láctico elevado em pacientes com GSD I. Quando os níveis de ácido láctico estão elevados, o ácido láctico transmitido pelo sangue compete pelo mesmo mecanismo de transporte tubular dos rins que o urato, limitando a taxa que o urato pode ser removido pelos rins para a urina. Se presente, o aumento do catabolismo purínico é um fator adicional que contribui para isso. Níveis de ácido úrico de 6 a 12 mg/dl (530 a 1060 umol/L) são comuns entre pacientes com GSD I, se a doença não for tratada adequadamente. Em algumas pessoas afetadas, o uso do medicamento allopurinol é necessário para diminuir os níveis de urina no sangue. As consequências da hiperuricemia entre pacientes com GSD I incluem o desenvolvimento de cálculos renais e o acúmulo de cristais de ácido úrico nas articulações, levando a doença renal e gota, respectivamente.
Hiperlipidemia e efeitos plasmáticosEditar
Triglicerídeos evitados no GSD I resultam de baixa insulina sérica em pacientes com hipoglicemia prolongada freqüente. Também pode ser causado pelo acúmulo intracelular de glucose-6-fosfato com derivação secundária para piruvato, que é convertido em Acetil-CoA, que é transportado para o citosol onde ocorre a síntese de ácidos graxos e colesterol. Triglicerídeos acima da faixa de 3,4 mmol/L (300 mg/dL) podem produzir lipemia visível, e até mesmo uma pseudo-hipponatremia leve devido a uma fração aquosa reduzida do plasma sanguíneo. No GSD I, o colesterol é tipicamente apenas levemente elevado quando comparado a outros lipídios.
HepatomegalyEdit
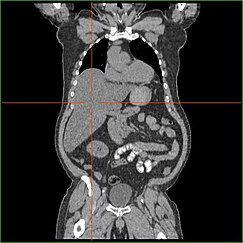
Impairment in the liver’s ability to perform gluconeogenesis leads to clinically apparent hepatomegaly. Sem este processo, o organismo é incapaz de libertar o glicogénio do fígado e convertê-lo em glicose sanguínea, levando a um acúmulo de glicogénio armazenado no fígado. A hepatomegalia do acúmulo de glicogênio armazenado no fígado é considerada uma forma de doença hepática gordurosa não-alcoólica. Os pacientes com GSD I apresentam um grau de hepatomegalia ao longo da vida, mas a gravidade muitas vezes está relacionada com o consumo de excesso de carboidratos dietéticos. Reduções na massa do fígado são possíveis, uma vez que a maioria dos pacientes retêm função hepática residual que permite a liberação de glicogênio armazenado a uma taxa limitada.
Pacientes com GSD I freqüentemente apresentam hepatomegalia desde o nascimento. No desenvolvimento fetal, a glicose materna transferida para o feto previne a hipoglicemia, mas o armazenamento da glicose como glicogênio no fígado leva à hepatomegalia. Não há evidências de que essa hepatomegalia apresente qualquer risco ao desenvolvimento fetal adequado.
Hepatomegalia em GSD tipo I geralmente ocorre sem o aumento simpático do baço. Os pacientes com GSD Ib podem apresentar esplenomegalia, mas isso está ligado ao uso de filgrastim para tratar a neutropenia nesse subtipo, não a hepatomegalia comorbida. A hepatomegalia persistirá até certo ponto ao longo da vida, causando frequentemente a protuberância do abdómen e, em casos graves, pode ser palpável no umbigo ou abaixo dele. Na doença hepática gordurosa não-alcoólica relacionada ao GSD, a função hepática é normalmente poupada, com enzimas hepáticas e bilirrubina permanecendo dentro da faixa normal. Entretanto, a função hepática pode ser afetada por outras complicações hepáticas na vida adulta, incluindo o desenvolvimento de adenomas hepáticos.
Adenomas hepáticosEditar
A etiologia específica dos adenomas hepáticos no GSD I permanece desconhecida, apesar das pesquisas em andamento. O paciente típico do GSD I que apresenta pelo menos um adenoma é um adulto, embora tenham sido observadas lesões em pacientes tão jovens quanto catorze anos. Adenomas, compostos de neoplasias heterogêneas, podem ocorrer individualmente ou em múltiplos. As estimativas sobre a taxa de conversão de um adenoma hepatocelular em carcinoma hepatocelular no GSD I variam de 0% a 11%, sendo que este último número representa pesquisas mais recentes. Uma razão para o aumento da estimativa é a crescente população de pacientes com GSD I sobrevivendo até a idade adulta, quando a maioria dos adenomas se desenvolve.
Os padrões de tratamento ditam a observação regular do fígado por RM ou tomografia computadorizada para monitorar anormalidades estruturais. Adenomas hepáticos podem ser mal identificados como hiperplasia nodular focal em imagens diagnósticas, embora esta condição seja rara. Entretanto, os adenomas hepáticos no GSD I envolvem exclusivamente deposição difusa de hialina de Mallory, o que é comumente observado na hiperplasia nodular focal. Ao contrário dos adenomas hepáticos comuns relacionados à contracepção oral, a hemorragia em pacientes com GSD I é rara.
Embora a razão para a alta prevalência de adenomas no GSD I não esteja clara, pesquisas desde os anos 70 têm implicado o glucágono sérico como um condutor potencial. Em estudos, pacientes que foram submetidos a um regime dietético para manter o açúcar no sangue em uma faixa normal de 72 a 108 mg/dL (4,0 a 6,0 mmol/L) têm mostrado uma diminuição na probabilidade de desenvolver adenomas. Além disso, pacientes com glicemia bem controlada têm visto consistentemente uma redução no tamanho e número de adenomas hepáticos, sugerindo que os adenomas podem ser causados por desequilíbrios de agentes hepatotrópicos como insulina sérica e especialmente glucagon sérico no fígado.
Hipertensão PortalEditar
OsteopeniaEdit
Patientes com GSD I desenvolverão frequentemente osteopenia. A etiologia específica da baixa densidade mineral óssea em GSD não é conhecida, embora esteja fortemente associada a um controle metabólico deficiente. A osteopenia pode ser causada diretamente pela hipoglicemia, ou pelas seqüelas endócrinas e metabólicas resultantes. As melhorias no controle metabólico têm demonstrado consistentemente prevenir ou reverter osteopenia clinicamente relevante em pacientes com GSD I. Nos casos em que a osteopenia progride com a idade, a densidade mineral óssea nas costelas é tipicamente mais grave do que nas vértebras. Em alguns casos, a densidade mineral óssea T-score cairá abaixo de -2,5, indicando osteoporose. Há algumas evidências de que a osteopenia pode estar ligada a anormalidades renais associadas no GSD I, particularmente a hiperfiltração glomular. A condição também parece reagir à suplementação de cálcio. Em muitos casos, a densidade mineral óssea pode aumentar e retornar à faixa normal, com controle metabólico adequado e suplementação de cálcio, revertendo a osteopenia.
Efeitos renaisEditar
Os rins costumam estar de 10 a 20% aumentados com glicogênio armazenado. Em adultos com GSD I, danos glomerulares crônicos semelhantes à nefropatia diabética podem levar à insuficiência renal. O GSD I pode apresentar várias complicações renais. Anomalias tubulares renais relacionadas à hiperlactatemia são vistas no início da vida, provavelmente porque a acidose láctica prolongada é mais provável de ocorrer na infância. Isto freqüentemente se apresentará como síndrome de Fanconi com múltiplas desarranjos de reabsorção tubular renal, incluindo acidose tubular com bicarbonato e desperdício de fosfato. Estas anomalias tubulares no GSD I são tipicamente detectadas e monitorizadas pelo cálcio urinário. A longo prazo, estas desarranjos podem exacerbar a nefropatia do ácido úrico, que de outra forma seria impulsionada pela hiperlactatemia. Na adolescência e além, a doença glomerular pode se desenvolver independentemente, inicialmente se apresentando como hiperfiltração glomerular indicada pela taxa de filtração glomerular elevada.
SplenomegalyEdit
Aumento do baço (esplenomegalia) é comum no GSD I e tem duas causas primárias. No GSD Ia, a esplenomegalia pode ser causada por uma relação entre o fígado e o baço, que faz com que cresça ou encolha para corresponder ao tamanho relativo do outro, a um grau menor. Em GSD Ib, é um efeito colateral do uso de filgrastim para tratar neutropenia.
Efeitos intestinaisEdit
Envolvimento intestinal pode causar má absorção com fezes gordurosas (esteatorreia), mas geralmente não requer tratamento.
Risco de infecçãoEdit
Neutropenia é uma característica distintiva de GSD Ib, ausente em GSD Ia. A causa microbiológica da neutropenia em GSD Ib não é bem compreendida. Em geral, o problema surge do metabolismo celular comprometido no neutrofilo, resultando na apoptose acelerada dos neutrófilos. A neutropenia no GSD é caracterizada tanto por uma diminuição da contagem absoluta de neutrófilos como pela diminuição da função dos neutrófilos. Os neutrófilos utilizam uma via metabólica específica do G6P que se baseia na presença de G6Pase-β ou G6PT para manter a homeostase energética dentro da célula. A ausência de G6PT no GSD Ib limita esta via, levando ao stress do retículo endoplasmático, stress oxidativo dentro do neutrofilo, desencadeando a apoptose prematura. O fator estimulante da colônia de granulócitos (G-CSF), disponível como filgrastim, pode reduzir o risco de infecção. Em alguns casos, o G-CSF formulado como pegfilgrastim, vendido sob o nome comercial Neulasta, pode ser usado como uma alternativa de ação lenta, requerendo dosagens menos freqüentes.
Trombocitopenia e problemas de coagulação sanguíneaEditar
A agregação plaquetária reparada é uma consequência incomum da hipoglicemia crônica, observada em pacientes com GSD I. Pesquisas demonstraram diminuição da função plaquetária, caracterizada por diminuição do consumo de protrombina, reações de agregação anormal, tempo de sangramento prolongado e baixa aderência plaquetária. A severidade da disfunção plaquetária geralmente se correlaciona com a condição clínica, com os casos mais graves se correlacionando com acidose láctica e lipidemia grave. Pode causar hemorragia clinicamente significativa, especialmente epistaxe. Além disso, pacientes com GSD I podem apresentar trombocitopenia como conseqüência de esplenomegalia. No cenário de esplenomegalia, vários fatores hematológicos podem ser seqüestrados nos tecidos do baço à medida que o sangue é filtrado através do órgão. Isto pode diminuir os níveis de plaquetas disponíveis na corrente sanguínea, levando à trombocitopenia.
Efeitos no desenvolvimentoEditar
O atraso no desenvolvimento é um potencial efeito secundário da hipoglicemia crônica ou recorrente, mas é, pelo menos teoricamente, evitável. As células neuronais e musculares normais não expressam glucose-6-fosfátase e, portanto, não são impactadas diretamente pelo GSD I. Entretanto, sem o tratamento adequado da hipoglicemia, a falha de crescimento geralmente resulta de níveis cronicamente baixos de insulina, acidose persistente, elevação crônica de hormônios catabólicos e insuficiência calórica (ou má absorção). Os atrasos de desenvolvimento mais dramáticos são frequentemente a causa de episódios graves (e não apenas persistentes) de hipoglicémia.