- Normal kulhydratbalance og opretholdelse af blodglukoseniveauetRediger
- PatofysiologiRediger
- Forhøjet laktat og mælkesyreacidoseRediger
- Forhøjet urat og komplikationerRediger
- Hyperlipidæmi og plasmaeffekterRediger
- HepatomegaliRediger
- LeveradenomerRediger
- Portal hypertensionRediger
- OsteopeniRediger
- NyrevirkningerRediger
- SplenomegaliRediger
- TarmeffekterRediger
- InfektionsrisikoRediger
- Trombocytopeni og problemer med blodkoagulationRediger
- Udviklingsmæssige virkningerRediger
Normal kulhydratbalance og opretholdelse af blodglukoseniveauetRediger
Glykogen i leveren og (i mindre grad) i nyrerne fungerer som en form for lagret, hurtigt tilgængeligt glukose, så blodglukoseniveauet kan opretholdes mellem måltiderne. I ca. 3 timer efter et kulhydratholdigt måltid styrer høje insulinniveauer levercellerne til at tage glukose fra blodet, til at omdanne det til glukose-6-fosfat (G6P) med enzymet glukokinase og til at tilføje G6P-molekylerne til enderne af glykogenkæderne (glykogensyntese). Overskydende G6P bliver også shuntet ind i produktionen af triglycerider og eksporteret til lagring i fedtvævet som fedt.
Når fordøjelsen af et måltid er afsluttet, falder insulinniveauet, og enzymsystemer i levercellerne begynder at fjerne glukosemolekyler fra glykogenstrenge i form af G6P. Denne proces betegnes glykogenolyse. G6P forbliver i levercellen, medmindre fosfatet spaltes af glukose-6-fosfatase. Denne dephosphoryleringsreaktion producerer fri glukose og frie PO
4-anioner. De frie glukosemolekyler kan transporteres ud af levercellerne til blodet for at opretholde en tilstrækkelig glukoseforsyning til hjernen og andre organer i kroppen. Glykogenolyse kan dække et voksent legemes glukosebehov i 12-18 timer.
Når fasten fortsætter i mere end et par timer, tillader faldende insulinniveauer katabolisme af muskelprotein og triglycerider fra fedtvævet. Produkterne fra disse processer er aminosyrer (hovedsagelig alanin), frie fedtsyrer og mælkesyre. Frie fedtsyrer fra triglycerider omdannes til ketoner og til acetyl-CoA. Aminosyrer og mælkesyre anvendes til at syntetisere nyt G6P i levercellerne ved glukoneogeneseprocessen. Det sidste trin i den normale glukoneogenese er ligesom det sidste trin i glykogenolysen defosforylering af G6P af glukose-6-fosfatase til fri glukose og PO
Dermed medierer glukose-6-fosfatase det sidste, centrale trin i begge de to hovedprocesser for glukoseproduktion under fastende. Effekten forstærkes, fordi de resulterende høje niveauer af glukose-6-fosfat hæmmer tidligere nøgletrin i både glykogenolyse og glukoneogenese.
PatofysiologiRediger
De vigtigste metaboliske virkninger af mangel på glukose-6-fosfatase er hypoglykæmi, mælkesyreacidose, hypertriglyceridæmi og hyperurikæmi.

Hypoglykæmien ved GSD I betegnes som “fastende” eller “post-absorptiv”, normalt omkring 4 timer efter fuldstændig fordøjelse af et måltid. Denne manglende evne til at opretholde et passende blodglukoseniveau under faste skyldes den kombinerede forringelse af både glykogenolyse og glukoneogenese. Fastehypoglykæmi er ofte det væsentligste problem ved GSD I og typisk det problem, der fører til diagnosen. Kronisk hypoglykæmi medfører sekundære metaboliske tilpasninger, herunder kronisk lave insulinniveauer og høje niveauer af glukagon og kortisol.
Lacetatacidose skyldes forringelse af glukoneogenesen. Mælkesyre dannes både i leveren og i musklerne og oxideres af NAD+ til pyrubrinsyre og omdannes derefter via den glukoneogene vej til G6P. Ophobning af G6P hæmmer omdannelsen af laktat til pyruvat. Mælkesyreniveauet stiger under faste i takt med, at glukosen falder. Hos personer med GSD I falder det måske ikke helt til det normale niveau, selv når normale glukoseniveauer genoprettes.
Hypertriglyceridæmi som følge af forstærket triglyceridproduktion er en anden indirekte virkning af nedsat glukoneogenese, der forstærkes af kronisk lave insulinniveauer. Under faste er den normale omdannelse af triglycerider til frie fedtsyrer, ketoner og i sidste ende acetyl-CoA forringet. Triglyceridniveauet i GSD I kan nå op på flere gange det normale niveau og tjener som et klinisk indeks for “metabolisk kontrol”.
Hyperurikæmi skyldes en kombination af øget dannelse og nedsat udskillelse af urinsyre, som dannes, når øgede mængder G6P metaboliseres via pentosephosphatvejen. Det er også et biprodukt af purinnedbrydning. Urinsyre konkurrerer med mælkesyre og andre organiske syrer om renal udskillelse i urinen. I GSD I medfører øget tilgængelighed af G6P til pentosephosphatvejen, øget katabolisme og nedsat urinudskillelse på grund af høje niveauer af mælkesyre tilsammen et urinsyreindhold flere gange højere end normalt. Selv om hyperurikæmi er asymptomatisk i årevis, opstår der gradvist skader på nyrer og led.
Forhøjet laktat og mælkesyreacidoseRediger
Høje niveauer af mælkesyre i blodet observeres hos alle personer med GSD I, som følge af nedsat glukoneogenese. Baselineforhøjninger varierer generelt fra 4 til 10 mol/mL, hvilket ikke vil medføre nogen kliniske konsekvenser. Under og efter en episode med lavt blodsukker vil laktatniveauerne imidlertid pludselig stige til over 15 mol/mL, som er tærsklen for mælkesyreacidose. Symptomer på laktacidose omfatter opkastning og hyperpnø, som begge kan forværre hypoglykæmi i forbindelse med GSD I. I tilfælde af akut laktacidose har patienterne brug for akut behandling for at stabilisere iltindholdet i blodet og genoprette blodglukose. Korrekt identifikation af mælkesyregigt hos børn, der ikke er diagnosticeret, er en udfordring, da de første symptomer typisk er opkastninger og dehydrering, som begge efterligner børneinfektioner som gastroenteritis eller pneumoni. Desuden kan begge disse almindelige infektioner fremskynde mere alvorlig hypoglykæmi hos udiagnosticerede børn, hvilket gør det vanskeligt at diagnosticere den underliggende årsag.
Da forhøjet laktat fortsætter, øger urinsyre, ketoacider og frie fedtsyrer aniongabet yderligere. Hos voksne og børn forårsager de høje koncentrationer af laktat et betydeligt ubehag i musklerne. Dette ubehag er en forstærket form af den brændende fornemmelse, som en løber kan føle i quadricepsmusklerne efter at have sprintet, og som skyldes en kortvarig ophobning af mælkesyre. Korrekt kontrol af hypoglykæmi i GSD I eliminerer helt muligheden for mælkesyreacidose.
Forhøjet urat og komplikationerRediger
Høje niveauer af urinsyre forekommer ofte som en konsekvens af forhøjet mælkesyre hos GSD I-patienter. Når laktatniveauerne er forhøjede, konkurrerer blodbåren mælkesyre om den samme nyretubulære transportmekanisme som urat, hvilket begrænser den hastighed, hvormed urat kan cleares af nyrerne til urinen. Hvis den er til stede, er øget purinkatabolisme en yderligere medvirkende faktor. Urinsyreniveauer på 6 til 12 mg/dl (530 til 1060 umol/L) er almindelige blandt GSD I-patienter, hvis sygdommen ikke behandles korrekt. Hos nogle af de berørte personer er det nødvendigt at anvende medicinen allopurinol for at sænke uratniveauet i blodet. Konsekvenserne af hyperurikæmi blandt GSD I-patienter omfatter udvikling af nyresten og ophobning af urinsyrekrystaller i leddene, hvilket fører til henholdsvis nyresygdom og gigt.
Hyperlipidæmi og plasmaeffekterRediger
Højere triglycerider hos GSD I skyldes lavt seruminsulin hos patienter med hyppig langvarig hypoglykæmi. Det kan også skyldes intracellulær ophobning af glukose-6-fosfat med sekundær shuntning til pyruvat, som omdannes til acetyl-CoA, der transporteres til cytosolen, hvor syntesen af fedtsyrer og kolesterol finder sted. Triglycerider over 3,4 mmol/L (300 mg/dL) kan give synlig lipæmi og endda en mild pseudohyponatriæmi på grund af en reduceret vandig fraktion i blodplasmaet. Ved GSD I er kolesterol typisk kun let forhøjet sammenlignet med andre lipider.
HepatomegaliRediger
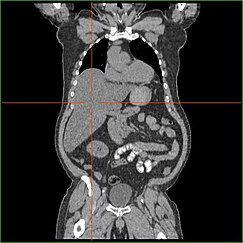
Svækkelse af leverens evne til at udføre glukoneogenese fører til klinisk synlig hepatomegali. Uden denne proces er kroppen ikke i stand til at frigøre glykogen fra leveren og omdanne det til blodglukose, hvilket fører til en ophobning af lagret glykogen i leveren. Hepatomegali som følge af ophobning af oplagret glykogen i leveren betragtes som en form for ikke-alkoholisk fedtleversygdom. GSD I-patienter har en vis grad af hepatomegali hele livet igennem, men sværhedsgraden hænger ofte sammen med indtagelse af overskydende kulhydrater i kosten. Reduktion af levermassen er mulig, da de fleste patienter bevarer en residual leverfunktion, der tillader frigørelse af oplagret glykogen i begrænset omfang.
GSD I-patienter har ofte hepatomegali fra fødslen. I den føtale udvikling forhindrer moderens glukose, der overføres til fosteret, hypoglykæmi, men lagring af glukose som glykogen i leveren fører til hepatomegali. Der er ingen beviser for, at denne hepatomegali udgør nogen risiko for en korrekt fosterudvikling.
Hepatomegali ved GSD type I forekommer generelt uden sympatisk forstørrelse af milten. GSD Ib-patienter kan præsentere sig med miltomegali, men dette hænger sammen med brugen af filgrastim til behandling af neutropeni i denne undertype, ikke komorbid hepatomegali. Hepatomegali vil i et vist omfang bestå hele livet igennem, hvilket ofte får maven til at rage frem og i svære tilfælde kan den være palpabel ved eller under navlen. Ved GSD-relateret ikke-alkoholisk fedtleversygdom er leverfunktionen normalt skånet, idet leverenzymer og bilirubin forbliver inden for normalområdet. Leverfunktionen kan dog blive påvirket af andre leverkomplikationer i voksenalderen, herunder udvikling af leveradenomer.
LeveradenomerRediger
Den specifikke ætiologi af leveradenomer hos GSD I er fortsat ukendt, på trods af igangværende forskning. Den typiske GSD I-patient, der præsenterer mindst ét adenom, er en voksen, selv om læsioner er blevet observeret hos patienter så unge som 14 år. Adenomer, der består af heterogene neoplasmer, kan forekomme enkeltvis eller i flere eksemplarer. Anslåede tal for omdannelse af et hepatocellulært adenom til hepatocellulært karcinom i GSD I varierer fra 0 % til 11 %, hvor sidstnævnte tal repræsenterer nyere forskning. En af årsagerne til det stigende skøn er den voksende population af GSD I-patienter, der overlever op i voksenalderen, hvor de fleste adenomer udvikles.
Behandlingsstandarder dikterer regelmæssig observation af leveren ved hjælp af MRI- eller CT-scanning for at overvåge for strukturelle abnormiteter. Hepatiske adenomer kan fejlagtigt identificeres som fokal nodulær hyperplasi i diagnostisk billeddannelse, selv om denne tilstand er sjælden. Lepraadenomer i GSD I omfatter imidlertid entydigt diffuse Mallory hyalinaflejringer, som ellers almindeligvis observeres ved fokal nodulær hyperplasi. I modsætning til almindelige hepatiske adenomer relateret til oral prævention er blødning hos GSD I-patienter sjældne.
Mens årsagen til den høje prævalens af adenomer i GSD I er uklar, har forskning siden 1970’erne impliceret serumglucagon som en potentiel drivkraft. I undersøgelser har patienter, der er blevet sat på et diætregime for at holde blodsukkeret i et normalt interval, der spænder fra 72 til 108 mg/dL (4,0 til 6,0 mmol/L), vist en nedsat sandsynlighed for udvikling af adenomer. Desuden har patienter med velkontrolleret blodsukker konsekvent set en reduktion i størrelsen og antallet af hepatiske adenomer, hvilket tyder på, at adenomer kan være forårsaget af ubalancer af hepatotrope stoffer som seruminsulin og især serumglucagon i leveren.
Portal hypertensionRediger
OsteopeniRediger
Patienter med GSD I vil ofte udvikle osteopeni. Den specifikke ætiologi for lav knoglemineraltæthed i GSD er ikke kendt, selv om den er stærkt forbundet med dårlig metabolisk kontrol. Osteopeni kan være direkte forårsaget af hypoglykæmi eller af de deraf følgende endokrine og metaboliske følgevirkninger. Forbedringer af den metaboliske kontrol har konsekvent vist sig at kunne forebygge eller vende klinisk relevant osteopeni hos GSD I-patienter. I de tilfælde, hvor osteopeni udvikler sig med alderen, er knoglemineraltætheden i ribbenene typisk mere alvorlig end i ryghvirvlerne. I nogle tilfælde vil knoglemineraltæthedens T-score falde til under -2,5, hvilket indikerer osteoporose. Der er visse tegn på, at osteopeni kan være forbundet med associerede nyreabnormaliteter hos GSD I, især glomulær hyperfiltrering. Tilstanden synes også at reagere på calciumtilskud. I mange tilfælde kan knoglemineraltætheden øges og vende tilbage til normalområdet ved korrekt metabolisk kontrol og calciumtilskud alene, hvilket vender osteopeni.
NyrevirkningerRediger
Nyrerne er normalt 10 til 20 % forstørrede med oplagret glykogen. Hos voksne med GSD I kan kronisk glomerulær skade svarende til diabetisk nefropati føre til nyresvigt. GSD I kan vise sig med forskellige nyrekomplikationer. Nyretubulære abnormiteter i forbindelse med hyperlaktatæmi ses tidligt i livet, sandsynligvis fordi langvarig mælkesyregigt er mere sandsynligt at forekomme i barndommen. Dette vil ofte vise sig som Fanconis syndrom med flere forstyrrelser i den renale tubulære reabsorption, herunder tubulær acidose med bicarbonat- og fosfatspild. Disse tubulære abnormiteter i GSD I påvises og overvåges typisk ved hjælp af urinkalcium. På lang sigt kan disse forstyrrelser forværre urinsyre nephropathi, der ellers er drevet af hyperlaktatæmi. I ungdomsårene og derefter kan der uafhængigt heraf udvikles glomerulær sygdom, der i første omgang viser sig som glomerulær hyperfiltrering indikeret ved forhøjet eGFR i urinen.
SplenomegaliRediger
Udvidelse af milten (splenomegali) er almindelig hos GSD I og har to primære årsager. Hos GSD Ia kan splenomegali skyldes et forhold mellem leveren og milten, som bevirker, at den ene vokser eller skrumper, så den svarer til den andens relative størrelse, i mindre grad. I GSD Ib er det en bivirkning ved brug af filgrastim til behandling af neutropeni.
TarmeffekterRediger
Intestinal involvering kan forårsage mild malabsorption med fedtet afføring (steatorrhea), men kræver normalt ingen behandling.
InfektionsrisikoRediger
Neutropeni er et karakteristisk træk ved GSD Ib, fraværende i GSD Ia. Den mikrobiologiske årsag til neutropeni i GSD Ib er ikke velforstået. Generelt skyldes problemet en kompromitteret cellulær metabolisme i neutrofilen, hvilket resulterer i en accelereret neutrofil apoptose. Neutropeni i GSD er karakteriseret ved både et fald i det absolutte neutrofiltal og en nedsat neutrofil funktion. Neutrofiler anvender en specifik G6P-metabolisk vej, som er afhængig af tilstedeværelsen af G6Pase-β eller G6PT for at opretholde energihomeostase i cellen. Fraværet af G6PT i GSD Ib begrænser denne vej, hvilket fører til stress i det endoplasmatiske retikulum og oxidativt stress i neutrofilen, hvilket udløser for tidlig apoptose. Granulocyt-koloni-stimulerende faktor (G-CSF), der fås som filgrastim, kan reducere risikoen for infektion. I nogle tilfælde kan G-CSF formuleret som pegfilgrastim, der sælges under handelsnavnet Neulasta, anvendes som et langsomt virkende alternativ, der kræver mindre hyppig dosering.
Trombocytopeni og problemer med blodkoagulationRediger
Svækket trombocytaggregation er en ualmindelig konsekvens af kronisk hypoglykæmi, der ses hos GSD I-patienter. Forskning har påvist nedsat trombocytfunktion, karakteriseret ved nedsat protrombinforbrug, unormale aggregationsreaktioner, forlænget blødningstid og lav trombocytadhæsionsevne. Sværhedsgraden af trombocytdysfunktion korrelerer typisk med den kliniske tilstand, idet de mest alvorlige tilfælde korrelerer med mælkesyre og alvorlig lipidæmi. Det kan forårsage klinisk betydelig blødning, især epistaxis. Desuden kan GSD I-patienter præsentere sig med trombocytopeni som følge af splenomegali. I forbindelse med splenomegali kan forskellige hæmatologiske faktorer blive sequesteret i miltens væv, når blodet filtreres gennem dette organ. Dette kan mindske niveauet af trombocytter, der er tilgængelige i blodbanen, hvilket fører til trombocytopeni.
Udviklingsmæssige virkningerRediger
Udviklingsforsinkelse er en potentiel sekundær virkning af kronisk eller tilbagevendende hypoglykæmi, men kan i det mindste teoretisk set forebygges. Normale neuronale celler og muskelceller udtrykker ikke glukose-6-fosfatase og påvirkes således ikke direkte af GSD I. Uden korrekt behandling af hypoglykæmi er vækstsvigt imidlertid almindeligvis resultatet af kronisk lave insulinniveauer, vedvarende acidose, kronisk forhøjelse af kataboliske hormoner og kalorieinsufficiens (eller malabsorption). De mest dramatiske udviklingsforsinkelser er ofte årsagen til alvorlige (ikke blot vedvarende) episoder af hypoglykæmi.