- Normales Kohlenhydratgleichgewicht und Aufrechterhaltung des BlutzuckerspiegelsEdit
- PathophysiologieBearbeiten
- Erhöhte Laktat- und MilchsäurewerteBearbeiten
- Erhöhter Harnsäurespiegel und KomplikationenBearbeiten
- Hyperlipidämie und PlasmaeffekteEdit
- HepatomegalieBearbeiten
- LeberadenomeBearbeiten
- Portale HypertonieBearbeiten
- OsteopenieBearbeiten
- Auswirkungen auf die NierenBearbeiten
- SplenomegalieBearbeiten
- Auswirkungen auf den DarmEdit
- InfektionsrisikoEdit
- Thrombozytopenie und BlutgerinnungsproblemeEdit
- Auswirkungen auf die EntwicklungBearbeiten
Normales Kohlenhydratgleichgewicht und Aufrechterhaltung des BlutzuckerspiegelsEdit
Glykogen in der Leber und (in geringerem Maße) in den Nieren dient als eine Form von gespeicherter, schnell verfügbarer Glukose, so dass der Blutzuckerspiegel zwischen den Mahlzeiten aufrechterhalten werden kann. Etwa drei Stunden lang nach einer kohlenhydrathaltigen Mahlzeit veranlasst ein hoher Insulinspiegel die Leberzellen, Glukose aus dem Blut aufzunehmen, sie mit Hilfe des Enzyms Glucokinase in Glukose-6-Phosphat (G6P) umzuwandeln und die G6P-Moleküle an die Enden der Glykogenketten anzuhängen (Glykogensynthese). Überschüssiges G6P wird auch in die Produktion von Triglyceriden umgelenkt und zur Speicherung im Fettgewebe als Fett exportiert.
Wenn die Verdauung einer Mahlzeit abgeschlossen ist, sinkt der Insulinspiegel, und Enzymsysteme in den Leberzellen beginnen, Glukosemoleküle in Form von G6P von Glykogensträngen zu entfernen. Dieser Vorgang wird als Glykogenolyse bezeichnet. Das G6P verbleibt in der Leberzelle, es sei denn, das Phosphat wird durch Glucose-6-Phosphatase gespalten. Bei dieser Dephosphorylierungsreaktion entstehen freie Glukose und freie PO
4-Anionen. Die freien Glukosemoleküle können aus den Leberzellen in das Blut transportiert werden, um das Gehirn und andere Organe des Körpers ausreichend mit Glukose zu versorgen. Die Glykogenolyse kann den Glukosebedarf eines erwachsenen Körpers 12-18 Stunden lang decken.
Wenn das Fasten länger als ein paar Stunden andauert, ermöglicht der sinkende Insulinspiegel den Abbau von Muskelprotein und Triglyceriden aus dem Fettgewebe. Die Produkte dieser Prozesse sind Aminosäuren (hauptsächlich Alanin), freie Fettsäuren und Milchsäure. Freie Fettsäuren aus Triglyceriden werden in Ketone und Acetyl-CoA umgewandelt. Aminosäuren und Milchsäure werden zur Synthese von neuem G6P in den Leberzellen durch den Prozess der Gluconeogenese verwendet. Der letzte Schritt der normalen Glukoneogenese ist, wie der letzte Schritt der Glykogenolyse, die Dephosphorylierung von G6P durch Glukose-6-Phosphatase zu freier Glukose und PO
Die Glukose-6-Phosphatase vermittelt also den letzten, entscheidenden Schritt in den beiden Hauptprozessen der Glukoseproduktion beim Fasten. Der Effekt wird dadurch verstärkt, dass die daraus resultierenden hohen Glucose-6-Phosphat-Spiegel frühere Schlüsselschritte sowohl in der Glykogenolyse als auch in der Gluconeogenese hemmen.
PathophysiologieBearbeiten
Die wichtigsten metabolischen Auswirkungen eines Mangels an Glucose-6-Phosphatase sind Hypoglykämie, Laktatazidose, Hypertriglyceridämie und Hyperurikämie.

Die Hypoglykämie bei GSD I wird als „nüchtern“ oder „post-absorptiv“ bezeichnet und tritt in der Regel etwa 4 Stunden nach der vollständigen Verdauung einer Mahlzeit auf. Die Unfähigkeit, einen angemessenen Blutzuckerspiegel während des Fastens aufrechtzuerhalten, resultiert aus einer kombinierten Beeinträchtigung der Glykogenolyse und der Glukoneogenese. Die Nüchternhypoglykämie ist häufig das wichtigste Problem bei GSD I und führt in der Regel zur Diagnose. Eine chronische Hypoglykämie führt zu sekundären Stoffwechselanpassungen, einschließlich chronisch niedriger Insulinspiegel und hoher Glukagonund Cortisolspiegel.
Die Milchsäureazidose entsteht durch eine Beeinträchtigung der Glukoneogenese. Milchsäure entsteht sowohl in der Leber als auch im Muskel und wird durch NAD+ zu Brenztraubensäure oxidiert und dann über den gluconeogenen Weg in G6P umgewandelt. Die Anhäufung von G6P hemmt die Umwandlung von Laktat in Pyruvat. Der Milchsäurespiegel steigt beim Fasten an, wenn die Glukose sinkt. Bei Menschen mit GSD I sinkt er möglicherweise nicht vollständig auf den Normalwert, selbst wenn normale Glukosespiegel wiederhergestellt werden.
Eine weitere indirekte Auswirkung der gestörten Glukoneogenese, die durch chronisch niedrige Insulinspiegel verstärkt wird, ist die Hypertriglyzeridämie, die aus einer verstärkten Triglyzeridproduktion resultiert. Während des Fastens ist die normale Umwandlung von Triglyceriden in freie Fettsäuren, Ketone und schließlich Acetyl-CoA beeinträchtigt. Die Triglyceridwerte bei GSD I können ein Mehrfaches des Normalwerts erreichen und dienen als klinischer Index für die „Stoffwechselkontrolle“.
Hyperurikämie ist das Ergebnis einer Kombination aus erhöhter Bildung und verminderter Ausscheidung von Harnsäure, die entsteht, wenn erhöhte Mengen von G6P über den Pentosephosphatweg verstoffwechselt werden. Sie ist auch ein Nebenprodukt des Purinabbaus. Harnsäure konkurriert mit Milchsäure und anderen organischen Säuren um die renale Ausscheidung im Urin. Bei GSD I führen die erhöhte Verfügbarkeit von G6P für den Pentosephosphatweg, die erhöhten Abbauraten und die verringerte Urinausscheidung aufgrund der hohen Milchsäurekonzentration zu Harnsäurespiegeln, die ein Mehrfaches des Normalwerts betragen. Obwohl die Hyperurikämie jahrelang asymptomatisch ist, treten allmählich Nieren- und Gelenkschäden auf.
Erhöhte Laktat- und MilchsäurewerteBearbeiten
Hohe Milchsäurewerte im Blut werden bei allen Menschen mit GSD I aufgrund der gestörten Glukoneogenese beobachtet. Die Ausgangswerte liegen im Allgemeinen zwischen 4 und 10 mol/ml, was keine klinischen Auswirkungen hat. Während und nach einer Unterzuckerung steigt der Laktatspiegel jedoch abrupt auf über 15 mol/ml an, was den Schwellenwert für eine Laktatazidose darstellt. Zu den Symptomen der Laktatazidose gehören Erbrechen und Hyperpnoe, die beide eine Hypoglykämie bei GSD I verschlimmern können. Bei akuter Laktatazidose benötigen die Patienten eine Notfallversorgung, um den Blutsauerstoff zu stabilisieren und den Blutzucker wiederherzustellen. Die korrekte Erkennung einer Laktatazidose bei nicht diagnostizierten Kindern stellt eine Herausforderung dar, da die ersten Symptome typischerweise Erbrechen und Dehydratation sind, die beide Infektionen im Kindesalter wie Gastroenteritis oder Lungenentzündung imitieren. Darüber hinaus können diese beiden häufigen Infektionen bei nicht diagnostizierten Kindern eine schwerere Hypoglykämie auslösen, was die Diagnose der zugrundeliegenden Ursache erschwert.
Wenn erhöhtes Laktat bestehen bleibt, vergrößern Harnsäure, Ketosäuren und freie Fettsäuren die Anionenlücke weiter. Bei Erwachsenen und Kindern verursachen die hohen Laktatkonzentrationen erhebliche Beschwerden in den Muskeln. Diese Beschwerden sind eine verstärkte Form des brennenden Gefühls, das ein Läufer nach einem Sprint im Quadrizeps verspürt und das durch eine kurzzeitige Anhäufung von Milchsäure verursacht wird. Eine ordnungsgemäße Kontrolle der Hypoglykämie bei GSD I schließt die Möglichkeit einer Laktatazidose vollständig aus.
Erhöhter Harnsäurespiegel und KomplikationenBearbeiten
Erhöhte Harnsäurespiegel treten bei GSD I-Patienten häufig als Folge einer erhöhten Milchsäure auf. Wenn die Laktatwerte erhöht sind, konkurriert die Milchsäure im Blut mit demselben Transportmechanismus in den Nierentubuli wie Urat, wodurch die Geschwindigkeit, mit der Urat über die Nieren in den Urin ausgeschieden werden kann, begrenzt wird. Falls vorhanden, ist ein erhöhter Purinkatabolismus ein weiterer Faktor, der dazu beiträgt. Harnsäurespiegel von 6 bis 12 mg/dl (530 bis 1060 umol/L) sind bei GSD I-Patienten üblich, wenn die Krankheit nicht angemessen behandelt wird. Bei einigen Betroffenen ist die Einnahme des Medikaments Allopurinol erforderlich, um den Uratspiegel im Blut zu senken. Zu den Folgen der Hyperurikämie bei GSD I-Patienten gehören die Entwicklung von Nierensteinen und die Ansammlung von Harnsäurekristallen in den Gelenken, was zu Nierenerkrankungen bzw. Gicht führt.
Hyperlipidämie und PlasmaeffekteEdit
Erhöhte Triglyceride bei GSD I sind das Ergebnis eines niedrigen Seruminsulins bei Patienten mit häufigen anhaltenden Hypoglykämien. Sie können auch durch eine intrazelluläre Anhäufung von Glukose-6-Phosphat mit sekundärem Shunt zu Pyruvat verursacht werden, das in Acetyl-CoA umgewandelt wird, das in das Zytosol transportiert wird, wo die Synthese von Fettsäuren und Cholesterin stattfindet. Triglyceride über dem Bereich von 3,4 mmol/L (300 mg/dL) können zu einer sichtbaren Lipämie und sogar zu einer leichten Pseudohyponatriämie führen, da der wässrige Anteil des Blutplasmas reduziert ist. Bei GSD I ist Cholesterin im Vergleich zu anderen Lipiden typischerweise nur leicht erhöht.
HepatomegalieBearbeiten
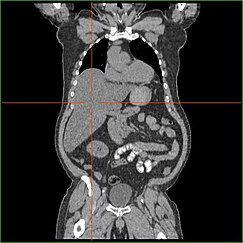
Eine Beeinträchtigung der Fähigkeit der Leber, Glukoneogenese zu betreiben, führt zu klinisch sichtbarer Hepatomegalie. Ohne diesen Prozess ist der Körper nicht in der Lage, Glykogen aus der Leber freizusetzen und in Blutglukose umzuwandeln, was zu einer Ansammlung von gespeichertem Glykogen in der Leber führt. Hepatomegalie aufgrund der Anhäufung von gespeichertem Glykogen in der Leber wird als eine Form der nichtalkoholischen Fettlebererkrankung angesehen. GSD I-Patienten weisen während ihres gesamten Lebens eine gewisse Hepatomegalie auf, deren Schweregrad jedoch häufig mit dem übermäßigen Verzehr von Kohlenhydraten in der Nahrung zusammenhängt. Eine Verringerung der Lebermasse ist möglich, da die meisten Patienten eine Restleberfunktion behalten, die die Freisetzung von gespeichertem Glykogen in begrenztem Umfang ermöglicht.
GSD I-Patienten weisen oft schon bei der Geburt eine Hepatomegalie auf. Während der fetalen Entwicklung verhindert die mütterliche Glukose, die auf den Fötus übertragen wird, eine Hypoglykämie, aber die Speicherung von Glukose als Glykogen in der Leber führt zu Hepatomegalie. Es gibt keine Hinweise darauf, dass diese Hepatomegalie ein Risiko für die ordnungsgemäße fetale Entwicklung darstellt.
Die Hepatomegalie bei GSD Typ I tritt im Allgemeinen ohne sympathische Vergrößerung der Milz auf. GSD-Ib-Patienten können eine Splenomegalie aufweisen, aber dies hängt mit der Verwendung von Filgrastim zur Behandlung der Neutropenie bei diesem Subtyp zusammen, nicht mit einer komorbiden Hepatomegalie. Die Hepatomegalie bleibt bis zu einem gewissen Grad lebenslang bestehen und führt häufig zu einer Vorwölbung des Bauches, die in schweren Fällen am oder unterhalb des Nabels tastbar sein kann. Bei der GSD-bedingten nicht-alkoholischen Fettlebererkrankung bleibt die Leberfunktion in der Regel erhalten, wobei die Leberenzyme und das Bilirubin im normalen Bereich liegen. Die Leberfunktion kann jedoch im Erwachsenenalter durch andere hepatische Komplikationen beeinträchtigt werden, einschließlich der Entwicklung von Leberadenomen.
LeberadenomeBearbeiten
Die spezifische Ätiologie von Leberadenomen bei GSD I ist trotz laufender Forschung noch immer unbekannt. Der typische GSD I-Patient, der mindestens ein Adenom aufweist, ist ein Erwachsener, obwohl Läsionen bei Patienten im Alter von vierzehn Jahren beobachtet wurden. Adenome, die sich aus heterogenen Neoplasmen zusammensetzen, können einzeln oder in Massen auftreten. Schätzungen über die Umwandlungsrate eines hepatozellulären Adenoms in ein hepatozelluläres Karzinom bei GSD I reichen von 0 % bis 11 %, wobei die letztere Zahl neueren Forschungen entspricht. Ein Grund für die steigende Schätzung ist die wachsende Zahl von GSD I-Patienten, die bis ins Erwachsenenalter überleben, in dem sich die meisten Adenome entwickeln.
Die Behandlungsstandards schreiben eine regelmäßige Beobachtung der Leber mittels MRT- oder CT-Scan vor, um strukturelle Anomalien festzustellen. Hepatische Adenome können in der diagnostischen Bildgebung fälschlicherweise als fokale noduläre Hyperplasie identifiziert werden, obwohl dieser Zustand selten ist. Hepatische Adenome bei GSD I weisen jedoch eindeutig eine diffuse Mallory-Hyalin-Ablagerung auf, die sonst häufig bei fokal-nodulärer Hyperplasie zu beobachten ist. Im Gegensatz zu häufigen hepatischen Adenomen im Zusammenhang mit oraler Kontrazeption sind Blutungen bei GSD I-Patienten selten.
Während der Grund für die hohe Prävalenz von Adenomen bei GSD I unklar ist, hat die Forschung seit den 1970er Jahren Serum-Glukagon als einen möglichen Auslöser in Betracht gezogen. In Studien zeigte sich, dass bei Patienten, die eine Diät einhalten mussten, um den Blutzuckerspiegel in einem normalen Bereich von 72 bis 108 mg/dL (4,0 bis 6,0 mmol/L) zu halten, die Wahrscheinlichkeit der Entwicklung von Adenomen geringer war. Darüber hinaus wurde bei Patienten mit gut kontrolliertem Blutzucker durchweg eine Verringerung der Größe und Anzahl von Leberadenomen festgestellt, was darauf hindeutet, dass Adenome durch ein Ungleichgewicht hepatotroper Substanzen wie Seruminsulin und insbesondere Serumglukagon in der Leber verursacht werden können.
Portale HypertonieBearbeiten
OsteopenieBearbeiten
Patienten mit GSD I entwickeln häufig eine Osteopenie. Die spezifische Ätiologie der niedrigen Knochenmineraldichte bei GSD ist nicht bekannt, obwohl sie stark mit einer schlechten Stoffwechseleinstellung verbunden ist. Die Osteopenie kann direkt durch Hypoglykämie oder die daraus resultierenden endokrinen und metabolischen Folgeerscheinungen verursacht werden. Eine Verbesserung der Stoffwechseleinstellung kann nachweislich eine klinisch relevante Osteopenie bei GSD I-Patienten verhindern oder umkehren. In Fällen, in denen die Osteopenie mit dem Alter fortschreitet, ist die Knochenmineraldichte in den Rippen typischerweise stärker ausgeprägt als in den Wirbeln. In einigen Fällen fällt der T-Score der Knochenmineraldichte unter -2,5, was auf Osteoporose hinweist. Es gibt Hinweise darauf, dass die Osteopenie mit den Nierenanomalien bei GSD I, insbesondere der glomulären Hyperfiltration, zusammenhängt. Die Erkrankung scheint auch auf eine Kalzium-Supplementierung anzusprechen. In vielen Fällen kann die Knochenmineraldichte zunehmen und in den Normalbereich zurückkehren, wenn eine angemessene Stoffwechselkontrolle und eine Kalziumergänzung durchgeführt werden, wodurch die Osteopenie rückgängig gemacht wird.
Auswirkungen auf die NierenBearbeiten
Die Nieren sind in der Regel um 10 bis 20 % vergrößert und enthalten Glykogen. Bei Erwachsenen mit GSD I kann eine chronische glomeruläre Schädigung ähnlich der diabetischen Nephropathie zu Nierenversagen führen. Bei GSD I können verschiedene Nierenkomplikationen auftreten. Nierentubuläre Anomalien im Zusammenhang mit Hyperlaktatämie treten schon früh im Leben auf, wahrscheinlich weil eine anhaltende Laktatazidose eher in der Kindheit auftritt. Dies äußert sich häufig als Fanconi-Syndrom mit multiplen Störungen der renalen tubulären Rückresorption, einschließlich tubulärer Azidose mit Bikarbonat- und Phosphatverlust. Diese tubulären Anomalien bei GSD I werden in der Regel durch Kalzium im Urin nachgewiesen und überwacht. Langfristig können diese Störungen die Harnsäurenephropathie verschlimmern, die ansonsten durch Hyperlaktatämie verursacht wird. In der Adoleszenz und darüber hinaus kann sich unabhängig davon eine glomeruläre Erkrankung entwickeln, die sich zunächst als glomeruläre Hyperfiltration zeigt und durch eine erhöhte Urin-EGFR angezeigt wird.
SplenomegalieBearbeiten
Eine Vergrößerung der Milz (Splenomegalie) ist bei GSD I häufig und hat zwei Hauptursachen. Bei der GSD Ia kann die Splenomegalie durch eine Beziehung zwischen Leber und Milz verursacht werden, die dazu führt, dass die eine Milz wächst oder schrumpft, um sich der relativen Größe der anderen anzugleichen, wenn auch in geringerem Maße. Bei GSD Ib ist sie eine Nebenwirkung der Verwendung von Filgrastim zur Behandlung der Neutropenie.
Auswirkungen auf den DarmEdit
Die Beteiligung des Darms kann zu einer leichten Malabsorption mit Fettstühlen (Steatorrhoe) führen, erfordert jedoch in der Regel keine Behandlung.
InfektionsrisikoEdit
Neutropenie ist ein charakteristisches Merkmal von GSD Ib, das bei GSD Ia fehlt. Die mikrobiologische Ursache der Neutropenie bei GSD Ib ist nicht gut verstanden. Im Großen und Ganzen ist das Problem auf einen gestörten Zellstoffwechsel der Neutrophilen zurückzuführen, der zu einer beschleunigten neutrophilen Apoptose führt. Die Neutropenie bei GSD ist sowohl durch eine Abnahme der absoluten Neutrophilenzahl als auch durch eine eingeschränkte Neutrophilenfunktion gekennzeichnet. Neutrophile nutzen einen spezifischen G6P-Stoffwechselweg, der auf das Vorhandensein von G6Pase-β oder G6PT angewiesen ist, um die Energiehomöostase innerhalb der Zelle aufrechtzuerhalten. Das Fehlen von G6PT bei GSD Ib schränkt diesen Stoffwechselweg ein, was zu Stress im endoplasmatischen Retikulum und oxidativem Stress innerhalb der Neutrophilen führt und eine vorzeitige Apoptose auslöst. Der Granulozyten-Kolonie-stimulierende Faktor (G-CSF), der als Filgrastim erhältlich ist, kann das Infektionsrisiko verringern. In einigen Fällen kann G-CSF in Form von Pegfilgrastim, das unter dem Handelsnamen Neulasta verkauft wird, als langsam wirkende Alternative verwendet werden, die eine weniger häufige Dosierung erfordert.
Thrombozytopenie und BlutgerinnungsproblemeEdit
Eine beeinträchtigte Thrombozytenaggregation ist eine seltene Folge der chronischen Hypoglykämie, die bei GSD I-Patienten beobachtet wird. In der Forschung wurde eine verminderte Thrombozytenfunktion nachgewiesen, die durch einen verminderten Prothrombinverbrauch, abnorme Aggregationsreaktionen, eine verlängerte Blutungszeit und eine geringe Thrombozytenadhäsion gekennzeichnet ist. Der Schweregrad der Thrombozytenfunktionsstörung korreliert typischerweise mit dem klinischen Zustand, wobei die schwersten Fälle mit Laktatazidose und schwerer Lipidämie korrelieren. Sie kann klinisch signifikante Blutungen, insbesondere Epistaxis, verursachen. Außerdem können GSD I-Patienten eine Thrombozytopenie als Folge einer Splenomegalie aufweisen. Bei einer Splenomegalie können verschiedene hämatologische Faktoren in den Geweben der Milz sequestriert werden, wenn das Blut durch das Organ gefiltert wird. Dies kann die Menge der im Blutkreislauf verfügbaren Blutplättchen verringern und zu Thrombozytopenie führen.
Auswirkungen auf die EntwicklungBearbeiten
Eine Entwicklungsverzögerung ist eine mögliche Nebenwirkung einer chronischen oder wiederkehrenden Hypoglykämie, ist aber zumindest theoretisch vermeidbar. Normale Neuronen- und Muskelzellen exprimieren keine Glukose-6-Phosphatase und werden daher nicht direkt von GSD I beeinträchtigt. Ohne eine angemessene Behandlung der Hypoglykämie kommt es jedoch häufig zu Wachstumsstörungen aufgrund chronisch niedriger Insulinspiegel, anhaltender Azidose, chronisch erhöhter kataboler Hormone und Kalorienmangel (oder Malabsorption). Die dramatischsten Entwicklungsverzögerungen sind oft die Ursache für schwere (nicht nur anhaltende) Hypoglykämie-Episoden.