- Equilibrio normal de los hidratos de carbono y mantenimiento de los niveles de glucosa en sangreEditar
- FisiopatologíaEditar
- Lactato elevado y acidosis lácticaEditar
- Urología elevada y complicacionesEditar
- Hiperlipidemia y efectos plasmáticosEditar
- HepatomegaliaEditar
- Adenomas hepáticosEditar
- Hipertensión portalEditar
- OsteopeniaEditar
- Efectos renalesEditar
- EsplenomegaliaEditar
- Efectos intestinalesEditar
- Riesgo de infecciónEditar
- Trombocitopenia y problemas de coagulación de la sangreEditar
- Efectos sobre el desarrolloEditar
Equilibrio normal de los hidratos de carbono y mantenimiento de los niveles de glucosa en sangreEditar
El glucógeno en el hígado y (en menor medida) en los riñones sirve como forma de glucosa almacenada y rápidamente accesible, de modo que el nivel de glucosa en sangre puede mantenerse entre las comidas. Durante unas 3 horas después de una comida que contenga carbohidratos, los altos niveles de insulina dirigen a las células del hígado para que tomen la glucosa de la sangre, la conviertan en glucosa-6-fosfato (G6P) con la enzima glucoquinasa y añadan las moléculas de G6P a los extremos de las cadenas de glucógeno (síntesis de glucógeno). El exceso de G6P también se desvía hacia la producción de triglicéridos y se exporta para su almacenamiento en el tejido adiposo en forma de grasa.
Cuando se completa la digestión de una comida, los niveles de insulina descienden y los sistemas enzimáticos de las células hepáticas comienzan a eliminar las moléculas de glucosa de las cadenas de glucógeno en forma de G6P. Este proceso se denomina glucogenolisis. El G6P permanece dentro de la célula hepática a menos que el fosfato sea escindido por la glucosa-6-fosfatasa. Esta reacción de desfosforilación produce glucosa libre y aniones PO
4 libres. Las moléculas de glucosa libre pueden ser transportadas fuera de las células hepáticas a la sangre para mantener un suministro adecuado de glucosa al cerebro y otros órganos del cuerpo. La glucogenólisis puede suplir las necesidades de glucosa de un cuerpo adulto durante 12-18 horas.
Cuando el ayuno se prolonga durante más de unas horas, el descenso de los niveles de insulina permite el catabolismo de las proteínas musculares y de los triglicéridos del tejido adiposo. Los productos de estos procesos son aminoácidos (principalmente alanina), ácidos grasos libres y ácido láctico. Los ácidos grasos libres de los triglicéridos se convierten en cetonas y en acetil-CoA. Los aminoácidos y el ácido láctico se utilizan para sintetizar nueva G6P en las células del hígado mediante el proceso de gluconeogénesis. El último paso de la gluconeogénesis normal, al igual que el último paso de la glucogenólisis, es la desfosforilación de la G6P por parte de la glucosa-6-fosfatasa para convertirla en glucosa libre y PO
Por lo tanto, la glucosa-6-fosfatasa media el paso final, clave, en ambos procesos principales de producción de glucosa durante el ayuno. El efecto se amplifica porque los altos niveles de glucosa-6-fosfato resultantes inhiben los pasos clave anteriores tanto en la glucogenolisis como en la gluconeogénesis.
FisiopatologíaEditar
Los principales efectos metabólicos de la deficiencia de glucosa-6-fosfatasa son la hipoglucemia, la acidosis láctica, la hipertrigliceridemia y la hiperuricemia.

La hipoglucemia de la GSD I se denomina «de ayuno», o «post-absorción», normalmente unas 4 horas después de la digestión completa de una comida. Esta incapacidad para mantener niveles adecuados de glucosa en sangre durante el ayuno es el resultado de la alteración combinada de la glucogenolisis y la gluconeogénesis. La hipoglucemia en ayunas suele ser el problema más importante en la EAG I, y suele ser el problema que conduce al diagnóstico. La hipoglucemia crónica produce adaptaciones metabólicas secundarias, incluyendo niveles de insulina crónicamente bajos y niveles elevados de glucagón y cortisol.
La acidosis láctica surge del deterioro de la gluconeogénesis. El ácido láctico se genera tanto en el hígado como en el músculo y es oxidado por el NAD+ a ácido pirúvico y luego convertido a través de la vía gluconeogénica a G6P. La acumulación de G6P inhibe la conversión de lactato en piruvato. El nivel de ácido láctico aumenta durante el ayuno a medida que la glucosa disminuye. En las personas con GSD I, es posible que no descienda por completo a la normalidad incluso cuando se restauran los niveles normales de glucosa.
La hipertrigliceridemia resultante de la producción amplificada de triglicéridos es otro efecto indirecto del deterioro de la gluconeogénesis, amplificado por los niveles crónicamente bajos de insulina. Durante el ayuno, la conversión normal de triglicéridos en ácidos grasos libres, cetonas y, en última instancia, acetil-CoA se ve afectada. Los niveles de triglicéridos en la EAG I pueden llegar a ser varias veces superiores a los normales y sirven como índice clínico de «control metabólico».
La hiperuricemia es el resultado de una combinación de aumento de la generación y disminución de la excreción de ácido úrico, que se genera cuando se metabolizan mayores cantidades de G6P a través de la vía de la pentosa fosfato. También es un subproducto de la degradación de las purinas. El ácido úrico compite con el ácido láctico y otros ácidos orgánicos por la excreción renal en la orina. En la EAG I, el aumento de la disponibilidad de G6P para la vía de las pentosas fosfato, el aumento de las tasas de catabolismo y la disminución de la excreción urinaria debido a los altos niveles de ácido láctico se combinan para producir niveles de ácido úrico varias veces superiores a los normales. Aunque la hiperuricemia es asintomática durante años, el daño renal y articular se acumula gradualmente.
Lactato elevado y acidosis lácticaEditar
Los niveles elevados de ácido láctico en la sangre se observan en todas las personas con GSD I, debido al deterioro de la gluconeogénesis. Las elevaciones basales suelen oscilar entre 4 y 10 mol/mL, lo que no causará ninguna repercusión clínica. Sin embargo, durante y después de un episodio de bajada de azúcar en sangre, los niveles de lactato aumentarán bruscamente hasta superar los 15 mol/mL, el umbral de la acidosis láctica. Los síntomas de la acidosis láctica incluyen vómitos e hiperpnea, que pueden exacerbar la hipoglucemia en el contexto de la EAG I. En los casos de acidosis láctica aguda, los pacientes necesitan atención de urgencia para estabilizar el oxígeno en sangre y restablecer la glucemia. La identificación adecuada de la acidosis láctica en niños no diagnosticados supone un reto, ya que los primeros síntomas suelen ser los vómitos y la deshidratación, ambos imitando infecciones infantiles como la gastroenteritis o la neumonía. Además, ambas infecciones comunes pueden precipitar una hipoglucemia más grave en niños no diagnosticados, lo que dificulta el diagnóstico de la causa subyacente.
Cuando persiste el lactato elevado, el ácido úrico, los cetoácidos y los ácidos grasos libres aumentan aún más la brecha aniónica. En los adultos y en los niños, las altas concentraciones de lactato provocan un importante malestar en los músculos. Estas molestias son una forma amplificada de la sensación de quemazón que un corredor puede sentir en los cuádriceps después de esprintar, causada por una breve acumulación de ácido láctico. El control adecuado de la hipoglucemia en la EAG I elimina por completo la posibilidad de que se produzca una acidosis láctica.
Urología elevada y complicacionesEditar
Los niveles elevados de ácido úrico suelen presentarse como consecuencia de la elevación del ácido láctico en los pacientes con EAG I. Cuando los niveles de lactato son elevados, el ácido láctico transportado por la sangre compite por el mismo mecanismo de transporte tubular renal que el urato, limitando la velocidad a la que el urato puede ser eliminado por los riñones en la orina. Si está presente, el aumento del catabolismo de las purinas es un factor adicional que contribuye. Los niveles de ácido úrico de 6 a 12 mg/dl (530 a 1060 umol/L) son comunes entre los pacientes con EAG I, si la enfermedad no se trata adecuadamente. En algunos afectados, es necesario el uso del medicamento alopurinol para reducir los niveles de ácido úrico en sangre. Las consecuencias de la hiperuricemia entre los pacientes con EAG I incluyen el desarrollo de cálculos renales y la acumulación de cristales de ácido úrico en las articulaciones, lo que conduce a la enfermedad renal y a la gota, respectivamente.
Hiperlipidemia y efectos plasmáticosEditar
La elevación de los triglicéridos en la EAG I es el resultado de una insulina sérica baja en pacientes con hipoglucemia prolongada frecuente. También puede estar causado por la acumulación intracelular de glucosa-6-fosfato con derivación secundaria a piruvato, que se convierte en Acetil-CoA, que es transportado al citosol donde se produce la síntesis de ácidos grasos y colesterol. Los triglicéridos por encima del rango de 3,4 mmol/L (300 mg/dL) pueden producir una lipemia visible, e incluso una leve pseudohiponatremia debido a la reducción de la fracción acuosa del plasma sanguíneo. En la EAG I, el colesterol suele estar ligeramente elevado en comparación con otros lípidos.
HepatomegaliaEditar
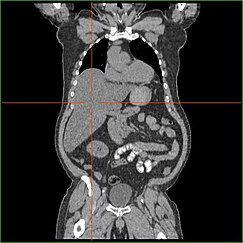
El deterioro de la capacidad del hígado para llevar a cabo la gluconeogénesis conduce a la hepatomegalia clínicamente aparente. Sin este proceso, el organismo es incapaz de liberar glucógeno del hígado y convertirlo en glucosa sanguínea, lo que conduce a una acumulación de glucógeno almacenado en el hígado. La hepatomegalia por acumulación de glucógeno almacenado en el hígado se considera una forma de enfermedad del hígado graso no alcohólico. Los pacientes con GSD I presentan cierto grado de hepatomegalia durante toda la vida, pero la gravedad suele estar relacionada con el consumo de un exceso de carbohidratos en la dieta. Es posible reducir la masa del hígado, ya que la mayoría de los pacientes conservan una función hepática residual que permite la liberación del glucógeno almacenado a un ritmo limitado.
Los pacientes con GSD I suelen presentar hepatomegalia desde el momento del nacimiento. En el desarrollo fetal, la glucosa materna transferida al feto evita la hipoglucemia, pero el almacenamiento de glucosa como glucógeno en el hígado conduce a la hepatomegalia. No hay pruebas de que esta hepatomegalia represente ningún riesgo para el correcto desarrollo del feto.
La hepatomegalia en la EAG tipo I se produce generalmente sin agrandamiento simpático del bazo. Los pacientes con EAG Ib pueden presentar esplenomegalia, pero esto está relacionado con el uso de filgrastim para tratar la neutropenia en este subtipo, no con la hepatomegalia comórbida. La hepatomegalia persistirá en cierto grado a lo largo de la vida, a menudo haciendo que el abdomen sobresalga, y en los casos graves puede ser palpable en el ombligo o por debajo de él. En la enfermedad del hígado graso no alcohólico relacionada con la EAG, la función hepática suele estar a salvo, y las enzimas hepáticas y la bilirrubina se mantienen dentro de los límites normales. Sin embargo, la función hepática puede verse afectada por otras complicaciones hepáticas en la edad adulta, incluido el desarrollo de adenomas hepáticos.
Adenomas hepáticosEditar
La etiología específica de los adenomas hepáticos en la EAG I sigue siendo desconocida, a pesar de las investigaciones en curso. El paciente típico de la EAG I que presenta al menos un adenoma es un adulto, aunque se han observado lesiones en pacientes de hasta catorce años. Los adenomas, compuestos por neoplasias heterogéneas, pueden aparecer individualmente o en múltiplos. Las estimaciones sobre la tasa de conversión de un adenoma hepatocelular en carcinoma hepatocelular en la EAG I oscilan entre el 0% y el 11%, siendo esta última cifra la que representa las investigaciones más recientes. Una de las razones del aumento de la estimación es la creciente población de pacientes con EAG I que sobreviven hasta la edad adulta, momento en el que se desarrollan la mayoría de los adenomas.
Las normas de tratamiento dictan la observación periódica del hígado mediante resonancia magnética o tomografía computarizada para controlar las anomalías estructurales. Los adenomas hepáticos pueden identificarse erróneamente como hiperplasia nodular focal en el diagnóstico por imagen, aunque esta condición es poco frecuente. Sin embargo, los adenomas hepáticos de la EAG I presentan de forma exclusiva una deposición hialina de Mallory difusa, que suele observarse en la hiperplasia nodular focal. A diferencia de los adenomas hepáticos comunes relacionados con los anticonceptivos orales, la hemorragia en los pacientes con GSD I es rara.
Aunque la razón de la alta prevalencia de adenomas en la GSD I no está clara, las investigaciones realizadas desde la década de 1970 han implicado al glucagón sérico como posible impulsor. En los estudios, los pacientes que han sido sometidos a un régimen dietético para mantener la glucemia en un rango normal que abarca de 72 a 108 mg/dL (4,0 a 6,0 mmol/L) han mostrado una menor probabilidad de desarrollar adenomas. Además, en los pacientes con la glucemia bien controlada se ha observado sistemáticamente una reducción del tamaño y el número de adenomas hepáticos, lo que sugiere que los adenomas pueden estar causados por desequilibrios de agentes hepatotrópicos como la insulina sérica y, especialmente, el glucagón sérico en el hígado.
Hipertensión portalEditar
OsteopeniaEditar
Los pacientes con EAG I suelen desarrollar osteopenia. No se conoce la etiología específica de la baja densidad mineral ósea en la EAG, aunque está fuertemente asociada a un mal control metabólico. La osteopenia puede estar causada directamente por la hipoglucemia o por las secuelas endocrinas y metabólicas resultantes. Las mejoras en el control metabólico han demostrado sistemáticamente que previenen o revierten la osteopenia clínicamente relevante en los pacientes con EAG I. En los casos en que la osteopenia progresa con la edad, la densidad mineral ósea en las costillas suele ser más grave que en las vértebras. En algunos casos, la puntuación T de la densidad mineral ósea caerá por debajo de -2,5, lo que indica osteoporosis. Hay algunas pruebas de que la osteopenia puede estar relacionada con las anomalías renales asociadas en la EAG I, en particular la hiperfiltración glomular. La enfermedad también parece responder a los suplementos de calcio. En muchos casos, la densidad mineral ósea puede aumentar y volver al rango normal si se realiza un control metabólico adecuado y se administra un suplemento de calcio, lo que revierte la osteopenia.
Efectos renalesEditar
Los riñones suelen estar agrandados en un 10 a 20% con glucógeno almacenado. En los adultos con GSD I, el daño glomerular crónico similar a la nefropatía diabética puede conducir a la insuficiencia renal. La EAG I puede presentar varias complicaciones renales. Las anomalías tubulares renales relacionadas con la hiperlactatemia se observan en las primeras etapas de la vida, probablemente porque es más probable que la acidosis láctica prolongada se produzca en la infancia. A menudo se presenta como un síndrome de Fanconi con múltiples alteraciones de la reabsorción tubular renal, incluida la acidosis tubular con pérdida de bicarbonato y fosfato. Estas anomalías tubulares en la EAG I suelen detectarse y controlarse por medio del calcio urinario. A largo plazo, estas alteraciones pueden exacerbar la nefropatía por ácido úrico, que por otra parte es provocada por la hiperlactatemia. A partir de la adolescencia, puede desarrollarse de forma independiente una enfermedad glomerular, que se presenta inicialmente como hiperfiltración glomerular indicada por una TFGe elevada en orina.
EsplenomegaliaEditar
El agrandamiento del bazo (esplenomegalia) es frecuente en la EAG I y tiene dos causas principales. En la EAG Ia, la esplenomegalia puede estar causada por una relación entre el hígado y el bazo que hace que cualquiera de los dos crezca o se encoja para igualar el tamaño relativo del otro, en un grado menor. En la EAG Ib, es un efecto secundario del uso de filgrastim para tratar la neutropenia.
Efectos intestinalesEditar
La afectación intestinal puede causar una malabsorción leve con heces grasientas (esteatorrea), pero normalmente no requiere tratamiento.
Riesgo de infecciónEditar
La neutropenia es una característica distintiva de la EAG Ib, ausente en la EAG Ia. La causa microbiológica de la neutropenia en la EAG Ib no se conoce bien. A grandes rasgos, el problema surge del compromiso del metabolismo celular de los neutrófilos, que da lugar a una apoptosis acelerada de los mismos. La neutropenia en la EAG se caracteriza tanto por la disminución del recuento absoluto de neutrófilos como por la disminución de su función. Los neutrófilos utilizan una vía metabólica específica de G6P que depende de la presencia de G6Pasa-β o G6PT para mantener la homeostasis energética dentro de la célula. La ausencia de G6PT en la EAG Ib limita esta vía, lo que conduce al estrés del retículo endoplásmico y al estrés oxidativo dentro del neutrófilo, desencadenando una apoptosis prematura. El factor estimulante de colonias de granulocitos (G-CSF), disponible como filgrastim, puede reducir el riesgo de infección. En algunos casos, el G-CSF formulado como pegfilgrastim, vendido bajo el nombre comercial de Neulasta, puede utilizarse como una alternativa de acción lenta, que requiere una dosificación menos frecuente.
Trombocitopenia y problemas de coagulación de la sangreEditar
La alteración de la agregación plaquetaria es una consecuencia poco frecuente de la hipoglucemia crónica, observada en pacientes con GSD I. Las investigaciones han demostrado una disminución de la función plaquetaria, caracterizada por una disminución del consumo de protrombina, reacciones de agregación anormales, tiempo de sangrado prolongado y baja adhesividad plaquetaria. La gravedad de la disfunción plaquetaria suele correlacionarse con el estado clínico, y los casos más graves se correlacionan con la acidosis láctica y la lipidemia grave. Puede causar hemorragias clínicamente significativas, especialmente epistaxis. Además, los pacientes con EAG I pueden presentar trombocitopenia como consecuencia de la esplenomegalia. En el contexto de la esplenomegalia, varios factores hematológicos pueden quedar secuestrados en los tejidos del bazo cuando la sangre se filtra a través del órgano. Esto puede disminuir los niveles de plaquetas disponibles en el torrente sanguíneo, dando lugar a trombocitopenia.
Efectos sobre el desarrolloEditar
El retraso del desarrollo es un efecto secundario potencial de la hipoglucemia crónica o recurrente, pero es al menos teóricamente prevenible. Las células neuronales y musculares normales no expresan glucosa-6-fosfatasa, por lo que no se ven afectadas por la EAG I directamente. Sin embargo, sin un tratamiento adecuado de la hipoglucemia, el fracaso del crecimiento suele ser consecuencia de los niveles crónicamente bajos de insulina, la acidosis persistente, la elevación crónica de las hormonas catabólicas y la insuficiencia calórica (o malabsorción). Los retrasos más dramáticos en el desarrollo suelen ser la causa de episodios graves (no sólo persistentes) de hipoglucemia.