- Equilibre glucidique normal et maintien de la glycémieModifié
- PathophysiologieModifier
- Lactate élevé et acidose lactiqueModifié
- Urate élevé et complicationsModifié
- Hyperlipidémie et effets plasmatiquesEdit
- HépatomégalieModifier
- Adénomes hépatiquesModifier
- Hypertension portaleModifier
- OstéopénieEdit
- Effets sur les reinsModifié
- SplénomégalieModifier
- Effets intestinauxEdit
- Risque infectieuxEdit
- Trombocytopénie et problèmes de coagulation sanguineModifier
- Effets sur le développementModifier
Equilibre glucidique normal et maintien de la glycémieModifié
Le glycogène dans le foie et (à un moindre degré) les reins sert de forme de glucose stocké, rapidement accessible, de sorte que la glycémie peut être maintenue entre les repas. Pendant environ 3 heures après un repas contenant des glucides, des taux élevés d’insuline incitent les cellules du foie à prélever le glucose dans le sang, à le convertir en glucose-6-phosphate (G6P) grâce à l’enzyme glucokinase, et à ajouter les molécules de G6P aux extrémités des chaînes de glycogène (synthèse du glycogène). L’excès de G6P est également dévié vers la production de triglycérides et exporté pour être stocké dans le tissu adipeux sous forme de graisse.
Lorsque la digestion d’un repas est terminée, les taux d’insuline chutent et les systèmes enzymatiques des cellules hépatiques commencent à éliminer les molécules de glucose des brins de glycogène sous forme de G6P. Ce processus est appelé glycogénolyse. Le G6P reste dans la cellule hépatique à moins que le phosphate ne soit clivé par la glucose-6-phosphatase. Cette réaction de déphosphorylation produit du glucose libre et des anions PO
4 libres. Les molécules de glucose libre peuvent être transportées hors des cellules hépatiques dans le sang pour maintenir un apport adéquat de glucose au cerveau et aux autres organes du corps. La glycogénolyse peut répondre aux besoins en glucose d’un corps adulte pendant 12 à 18 heures.
Lorsque le jeûne se poursuit pendant plus de quelques heures, la chute des taux d’insuline permet le catabolisme des protéines musculaires et des triglycérides du tissu adipeux. Les produits de ces processus sont des acides aminés (principalement l’alanine), des acides gras libres et de l’acide lactique. Les acides gras libres des triglycérides sont convertis en cétones et en acétyl-CoA. Les acides aminés et l’acide lactique sont utilisés pour synthétiser du nouveau G6P dans les cellules du foie par le processus de la gluconéogenèse. La dernière étape de la gluconéogenèse normale, comme la dernière étape de la glycogénolyse, est la déphosphorylation du G6P par la glucose-6-phosphatase en glucose libre et en PO
Donc la glucose-6-phosphatase est le médiateur de l’étape finale, clé, dans les deux principaux processus de production de glucose pendant le jeûne. L’effet est amplifié parce que les niveaux élevés de glucose-6-phosphate qui en résultent inhibent les étapes clés antérieures de la glycogénolyse et de la gluconéogenèse.
PathophysiologieModifier
Les principaux effets métaboliques de la déficience en glucose-6-phosphatase sont l’hypoglycémie, l’acidose lactique, l’hypertriglycéridémie et l’hyperuricémie.

L’hypoglycémie du GSD I est dite « à jeun », ou « post-absorptive », généralement environ 4 heures après la digestion complète d’un repas. Cette incapacité à maintenir une glycémie adéquate pendant le jeûne résulte de l’altération combinée de la glycogénolyse et de la néoglucogenèse. L’hypoglycémie de jeûne est souvent le problème le plus important dans la GSD I, et typiquement le problème qui conduit au diagnostic. L’hypoglycémie chronique produit des adaptations métaboliques secondaires, notamment des taux d’insuline chroniquement bas et des taux élevés de glucagon et de cortisol.
L’acidose lactique résulte d’une altération de la gluconéogenèse. L’acide lactique est généré à la fois dans le foie et dans les muscles et est oxydé par le NAD+ en acide pyruvique, puis converti en G6P via la voie gluconéogénique. L’accumulation de G6P inhibe la conversion du lactate en pyruvate. Le taux d’acide lactique augmente pendant le jeûne alors que le glucose diminue. Chez les personnes atteintes de GSD I, il peut ne pas retomber entièrement à la normale même lorsque les taux de glucose normaux sont rétablis.
L’hypertriglycéridémie résultant d’une production amplifiée de triglycérides est un autre effet indirect de l’altération de la gluconéogenèse, amplifiée par des taux d’insuline chroniquement bas. Pendant le jeûne, la conversion normale des triglycérides en acides gras libres, en cétones et, finalement, en acétyl-CoA est altérée. Les niveaux de triglycérides dans le GSD I peuvent atteindre plusieurs fois la normale et servent d’indice clinique de « contrôle métabolique ».
L’hyperuricémie résulte de la combinaison d’une augmentation de la génération et d’une diminution de l’excrétion de l’acide urique, qui est généré lorsque des quantités accrues de G6P sont métabolisées via la voie des pentoses phosphates. Il s’agit également d’un sous-produit de la dégradation des purines. L’acide urique est en compétition avec l’acide lactique et d’autres acides organiques pour l’excrétion rénale dans l’urine. Dans la GSD I, la disponibilité accrue de G6P pour la voie des pentoses phosphates, l’augmentation des taux de catabolisme et la diminution de l’excrétion urinaire due à des niveaux élevés d’acide lactique se combinent pour produire des niveaux d’acide urique plusieurs fois supérieurs à la normale. Bien que l’hyperuricémie soit asymptomatique pendant des années, les dommages rénaux et articulaires s’accumulent progressivement.
Lactate élevé et acidose lactiqueModifié
Des niveaux élevés d’acide lactique dans le sang sont observés chez toutes les personnes atteintes de GSD I, en raison de l’altération de la gluconéogenèse. Les élévations de base se situent généralement entre 4 et 10 mol/mL, ce qui n’entraînera aucun impact clinique. Cependant, pendant et après un épisode d’hypoglycémie, le taux de lactate augmente brusquement pour dépasser 15 mol/mL, seuil de l’acidose lactique. Les symptômes de l’acidose lactique comprennent les vomissements et l’hyperpnée, qui peuvent tous deux exacerber l’hypoglycémie dans le cadre de la GSD I. En cas d’acidose lactique aiguë, les patients ont besoin de soins d’urgence pour stabiliser l’oxygène sanguin et rétablir la glycémie. Il est difficile d’identifier correctement l’acidose lactique chez les enfants non diagnostiqués, car les premiers symptômes sont généralement des vomissements et une déshydratation, qui ressemblent tous deux à des infections infantiles comme la gastro-entérite ou la pneumonie. De plus, ces deux infections courantes peuvent précipiter une hypoglycémie plus sévère chez les enfants non diagnostiqués, ce qui rend le diagnostic de la cause sous-jacente difficile.
Lorsque le lactate élevé persiste, l’acide urique, les cétoacides et les acides gras libres augmentent encore le trou anionique. Chez les adultes et les enfants, les concentrations élevées de lactate provoquent un inconfort important dans les muscles. Cette gêne est une forme amplifiée de la sensation de brûlure qu’un coureur peut ressentir dans les quadriceps après un sprint, qui est causée par une brève accumulation d’acide lactique. Un contrôle approprié de l’hypoglycémie dans le GSD I élimine entièrement la possibilité d’une acidose lactique.
Urate élevé et complicationsModifié
Des niveaux élevés d’acide urique se présentent souvent comme une conséquence de l’acide lactique élevé chez les patients GSD I. Lorsque les taux de lactate sont élevés, l’acide lactique véhiculé par le sang entre en compétition pour le même mécanisme de transport tubulaire rénal que l’urate, ce qui limite la vitesse à laquelle l’urate peut être éliminé par les reins dans l’urine. S’il est présent, l’augmentation du catabolisme des purines est un facteur supplémentaire. Des taux d’acide urique de 6 à 12 mg/dl (530 à 1060 umol/L) sont courants chez les patients atteints de GSD I, si la maladie n’est pas correctement traitée. Chez certaines personnes atteintes, l’utilisation du médicament allopurinol est nécessaire pour abaisser le taux d’urate sanguin. Les conséquences de l’hyperuricémie chez les patients atteints de GSD I comprennent le développement de calculs rénaux et l’accumulation de cristaux d’acide urique dans les articulations, conduisant respectivement à une maladie rénale et à la goutte.
Hyperlipidémie et effets plasmatiquesEdit
L’élévation des triglycérides chez les patients atteints de GSD I résulte d’un faible taux d’insuline sérique chez les patients présentant des hypoglycémies prolongées fréquentes. Elle peut également être causée par une accumulation intracellulaire de glucose-6-phosphate avec un shuntage secondaire en pyruvate, qui est converti en Acétyl-CoA, lequel est transporté vers le cytosol où se produit la synthèse des acides gras et du cholestérol. Des triglycérides supérieurs à 3,4 mmol/L (300 mg/dL) peuvent produire une lipémie visible, voire une légère pseudo-hyponatrémie due à une fraction aqueuse réduite du plasma sanguin. Dans le GSD I, le cholestérol n’est généralement que légèrement élevé par rapport aux autres lipides.
HépatomégalieModifier
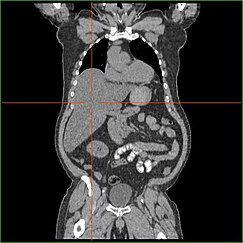
L’altération de la capacité du foie à effectuer la gluconéogenèse entraîne une hépatomégalie cliniquement apparente. Sans ce processus, l’organisme est incapable de libérer le glycogène du foie et de le convertir en glucose sanguin, ce qui entraîne une accumulation de glycogène stocké dans le foie. L’hépatomégalie due à l’accumulation de glycogène stocké dans le foie est considérée comme une forme de stéatose hépatique non alcoolique. Les patients atteints de GSD I présentent un certain degré d’hépatomégalie tout au long de leur vie, mais la gravité est souvent liée à la consommation d’un excès de glucides alimentaires. Des réductions de la masse du foie sont possibles, car la plupart des patients conservent une fonction hépatique résiduelle qui permet la libération du glycogène stocké à un rythme limité.
Les patients GSD I présentent souvent une hépatomégalie dès la naissance. Au cours du développement fœtal, le glucose maternel transféré au fœtus empêche l’hypoglycémie, mais le stockage du glucose sous forme de glycogène dans le foie entraîne une hépatomégalie. Il n’y a aucune preuve que cette hépatomégalie présente un risque pour le bon développement du fœtus.
L’hépatomégalie dans la GSD de type I se produit généralement sans hypertrophie sympathique de la rate. Les patients GSD Ib peuvent présenter une splénomégalie, mais cela est lié à l’utilisation du filgrastim pour traiter la neutropénie dans ce sous-type, et non à une hépatomégalie comorbide. L’hépatomégalie persistera à un certain degré tout au long de la vie, provoquant souvent une protubérance de l’abdomen et, dans les cas graves, elle peut être palpable au niveau ou en dessous du nombril. Dans la stéatose hépatique non alcoolique liée à la GSD, la fonction hépatique est généralement épargnée, les enzymes hépatiques et la bilirubine restant dans les limites de la normale. Cependant, la fonction hépatique peut être affectée par d’autres complications hépatiques à l’âge adulte, notamment le développement d’adénomes hépatiques.
Adénomes hépatiquesModifier
L’étiologie spécifique des adénomes hépatiques dans la GSD I reste inconnue, malgré les recherches en cours. Le patient typique du GSD I présentant au moins un adénome est un adulte, bien que des lésions aient été observées chez des patients âgés de quatorze ans seulement. Les adénomes, composés de néoplasmes hétérogènes, peuvent apparaître individuellement ou en multiples. Les estimations du taux de conversion d’un adénome hépatocellulaire en carcinome hépatocellulaire dans le GSD I vont de 0 à 11 %, ce dernier chiffre représentant les recherches les plus récentes. Une des raisons de l’augmentation de l’estimation est la population croissante de patients atteints de GSD I qui survivent jusqu’à l’âge adulte, lorsque la plupart des adénomes se développent.
Les normes de traitement dictent une observation régulière du foie par IRM ou tomodensitométrie pour surveiller les anomalies structurelles. Les adénomes hépatiques peuvent être identifiés à tort comme une hyperplasie nodulaire focale lors de l’imagerie diagnostique, bien que cette condition soit rare. Cependant, les adénomes hépatiques dans la GSD I impliquent uniquement un dépôt hyalin diffus de Mallory, qui est autrement couramment observé dans l’hyperplasie nodulaire focale. Contrairement aux adénomes hépatiques courants liés à la contraception orale, l’hémorragie chez les patients atteints de GSD I est rare.
Bien que la raison de la prévalence élevée des adénomes dans le GSD I ne soit pas claire, la recherche depuis les années 1970 a impliqué le glucagon sérique comme un conducteur potentiel. Dans les études, les patients qui ont été soumis à un régime alimentaire pour maintenir la glycémie dans une fourchette normale de 72 à 108 mg/dL (4,0 à 6,0 mmol/L) ont montré une probabilité réduite de développer des adénomes. De plus, les patients dont la glycémie est bien contrôlée ont constamment vu une réduction de la taille et du nombre d’adénomes hépatiques, ce qui suggère que les adénomes peuvent être causés par des déséquilibres des agents hépatotropes comme l’insuline sérique et surtout le glucagon sérique dans le foie.
Hypertension portaleModifier
OstéopénieEdit
Les patients atteints de GSD I développeront souvent une ostéopénie. L’étiologie spécifique de la faible densité minérale osseuse dans la GSD n’est pas connue, bien qu’elle soit fortement associée à un mauvais contrôle métabolique. L’ostéopénie peut être directement causée par l’hypoglycémie, ou par les séquelles endocriniennes et métaboliques qui en résultent. Il a toujours été démontré que l’amélioration du contrôle métabolique permettait de prévenir ou d’inverser l’ostéopénie cliniquement pertinente chez les patients atteints de GSD I. Dans les cas où l’ostéopénie progresse avec l’âge, la densité minérale osseuse dans les côtes est généralement plus sévère que dans les vertèbres. Dans certains cas, le score T de la densité minérale osseuse tombe en dessous de -2,5, ce qui indique une ostéoporose. Il semble que l’ostéopénie puisse être liée à des anomalies rénales associées dans la GSD I, en particulier l’hyperfiltration glomulaire. Cette affection semble également répondre à une supplémentation en calcium. Dans de nombreux cas, la densité minérale osseuse peut augmenter et revenir à la normale compte tenu d’un contrôle métabolique approprié et d’une supplémentation en calcium seule, inversant l’ostéopénie.
Effets sur les reinsModifié
Les reins sont généralement hypertrophiés de 10 à 20 % avec le glycogène stocké. Chez les adultes atteints de GSD I, des dommages glomérulaires chroniques similaires à la néphropathie diabétique peuvent conduire à une insuffisance rénale. Le GSD I peut présenter diverses complications rénales. Les anomalies tubulaires rénales liées à l’hyperlactatémie sont observées tôt dans la vie, probablement parce que l’acidose lactique prolongée est plus susceptible de se produire dans l’enfance. Ces anomalies se présentent souvent sous la forme d’un syndrome de Fanconi avec de multiples dérèglements de la réabsorption tubulaire rénale, y compris une acidose tubulaire avec perte de bicarbonate et de phosphate. Ces anomalies tubulaires dans la GSD I sont typiquement détectées et surveillées par le calcium urinaire. À long terme, ces dérèglements peuvent exacerber la néphropathie à acide urique, autrement provoquée par l’hyperlactatémie. À l’adolescence et au-delà, une maladie glomérulaire peut se développer indépendamment, se présentant initialement comme une hyperfiltration glomérulaire indiquée par un DFGe urinaire élevé.
SplénomégalieModifier
L’hypertrophie de la rate (splénomégalie) est fréquente dans la GSD I et a deux causes principales. Dans le GSD Ia, la splénomégalie peut être causée par une relation entre le foie et la rate qui fait que l’un ou l’autre grandit ou rétrécit pour correspondre à la taille relative de l’autre, à un degré moindre. Dans le GSD Ib, c’est un effet secondaire de l’utilisation du filgrastim pour traiter la neutropénie.
Effets intestinauxEdit
L’atteinte intestinale peut provoquer une légère malabsorption avec des selles grasses (stéatorrhée), mais ne nécessite généralement pas de traitement.
Risque infectieuxEdit
La neutropénie est une caractéristique distinctive du GSD Ib, absente dans le GSD Ia. La cause microbiologique de la neutropénie dans le GSD Ib n’est pas bien comprise. En gros, le problème découle d’un métabolisme cellulaire compromis dans le neutrophile, ce qui entraîne une apoptose accélérée du neutrophile. La neutropénie dans la GSD se caractérise à la fois par une diminution du nombre absolu de neutrophiles et par une diminution de la fonction neutrophile. Les neutrophiles utilisent une voie métabolique spécifique du G6P qui repose sur la présence de la G6Pase-β ou G6PT pour maintenir l’homéostasie énergétique au sein de la cellule. L’absence de G6PT dans la GSD Ib limite cette voie, ce qui entraîne un stress du réticulum endoplasmique, un stress oxydatif au sein du neutrophile, déclenchant une apoptose prématurée. Le facteur de stimulation des colonies de granulocytes (G-CSF), disponible sous forme de filgrastim, peut réduire le risque d’infection. Dans certains cas, le G-CSF formulé sous forme de pegfilgrastim, vendu sous le nom commercial Neulasta, peut être utilisé comme une alternative à action lente, nécessitant des doses moins fréquentes.
Trombocytopénie et problèmes de coagulation sanguineModifier
L’altération de l’agrégation plaquettaire est une conséquence peu fréquente de l’hypoglycémie chronique, observée chez les patients GSD I. La recherche a démontré une diminution de la fonction plaquettaire, caractérisée par une diminution de la consommation de prothrombine, des réactions d’agrégation anormales, un temps de saignement prolongé et une faible adhésivité plaquettaire. La gravité du dysfonctionnement plaquettaire est généralement en corrélation avec l’état clinique, les cas les plus graves étant en corrélation avec l’acidose lactique et la lipidémie sévère. Elle peut provoquer des saignements cliniquement significatifs, notamment des épistaxis. De plus, les patients atteints de GSD I peuvent présenter une thrombocytopénie comme conséquence de la splénomégalie. Dans le cadre d’une splénomégalie, divers facteurs hématologiques peuvent être séquestrés dans les tissus de la rate lorsque le sang est filtré par cet organe. Cela peut diminuer les niveaux de plaquettes disponibles dans la circulation sanguine, entraînant une thrombocytopénie.
Effets sur le développementModifier
Le retard de développement est un effet secondaire potentiel de l’hypoglycémie chronique ou récurrente, mais il est au moins théoriquement évitable. Les cellules neuronales et musculaires normales n’expriment pas la glucose-6-phosphatase, et ne sont donc pas impactées directement par la GSD I. Cependant, en l’absence d’un traitement approprié de l’hypoglycémie, le retard de croissance résulte généralement de taux d’insuline chroniquement bas, d’une acidose persistante, d’une élévation chronique des hormones cataboliques et d’une insuffisance calorique (ou malabsorption). Les retards de développement les plus spectaculaires sont souvent la cause d’épisodes graves (et pas seulement persistants) d’hypoglycémie.