- Normale equilibrio dei carboidrati e mantenimento dei livelli di glucosio nel sangueModifica
- FisiopatologiaModifica
- Lattato elevato e acidosi latticaModifica
- Urati elevati e complicazioniModifica
- Iperlipidemia ed effetti plasmaticiModifica
- EpatomegaliaModifica
- Adenomi epaticiModifica
- Ipertensione portaleModifica
- OsteopeniaModifica
- Effetti renaliModifica
- SplenomegaliaModifica
- Effetti intestinaliModifica
- Rischio di infezioneModifica
- Trombocitopenia e problemi di coagulazione del sangueModifica
- Effetti sullo sviluppoModifica
Normale equilibrio dei carboidrati e mantenimento dei livelli di glucosio nel sangueModifica
Il glicogeno nel fegato e (in misura minore) nei reni serve come forma di glucosio immagazzinato e rapidamente accessibile, in modo che il livello di glucosio nel sangue possa essere mantenuto tra i pasti. Per circa 3 ore dopo un pasto contenente carboidrati, alti livelli di insulina dirigono le cellule epatiche a prendere il glucosio dal sangue, a convertirlo in glucosio-6-fosfato (G6P) con l’enzima glucochinasi, e ad aggiungere le molecole di G6P alle estremità delle catene di glicogeno (sintesi del glicogeno). Il G6P in eccesso viene anche deviato nella produzione di trigliceridi ed esportato per essere immagazzinato nel tessuto adiposo come grasso.
Quando la digestione di un pasto è completa, i livelli di insulina scendono e i sistemi enzimatici nelle cellule epatiche iniziano a rimuovere le molecole di glucosio dalle catene di glicogeno sotto forma di G6P. Questo processo è chiamato glicogenolisi. Il G6P rimane all’interno della cellula epatica a meno che il fosfato non venga scisso dalla glucosio-6-fosfatasi. Questa reazione di defosforilazione produce glucosio libero e anioni PO
4 liberi. Le molecole di glucosio libero possono essere trasportate fuori dalle cellule epatiche nel sangue per mantenere un adeguato apporto di glucosio al cervello e ad altri organi del corpo. La glicogenolisi può fornire il fabbisogno di glucosio di un corpo adulto per 12-18 ore.
Quando il digiuno continua per più di qualche ora, il calo dei livelli di insulina permette il catabolismo delle proteine muscolari e dei trigliceridi del tessuto adiposo. I prodotti di questi processi sono aminoacidi (principalmente alanina), acidi grassi liberi e acido lattico. Gli acidi grassi liberi dai trigliceridi sono convertiti in chetoni e in acetil-CoA. Gli amminoacidi e l’acido lattico sono usati per sintetizzare nuovo G6P nelle cellule epatiche attraverso il processo di gluconeogenesi. L’ultimo passo della gluconeogenesi normale, come l’ultimo passo della glicogenolisi, è la defosforilazione del G6P da parte della glucosio-6-fosfatasi in glucosio libero e PO
Quindi la glucosio-6-fosfatasi media il passo finale, chiave, in entrambi i due processi principali di produzione del glucosio durante il digiuno. L’effetto è amplificato perché gli alti livelli risultanti di glucosio-6-fosfato inibiscono i precedenti passaggi chiave sia nella glicogenolisi che nella gluconeogenesi.
FisiopatologiaModifica
I principali effetti metabolici della carenza di glucosio-6-fosfatasi sono ipoglicemia, acidosi lattica, ipertrigliceridemia e iperuricemia.

L’ipoglicemia della GSD I è definita “a digiuno”, o “post-assorbitiva”, solitamente circa 4 ore dopo la completa digestione di un pasto. Questa incapacità di mantenere adeguati livelli di glucosio nel sangue durante il digiuno deriva dalla compromissione combinata della glicogenolisi e della gluconeogenesi. L’ipoglicemia a digiuno è spesso il problema più significativo nella GSD I, e tipicamente il problema che porta alla diagnosi. L’ipoglicemia cronica produce adattamenti metabolici secondari, tra cui livelli di insulina cronicamente bassi e alti livelli di glucagone e cortisolo.
L’acidosi lattica deriva dalla compromissione della gluconeogenesi. L’acido lattico è generato sia nel fegato che nel muscolo ed è ossidato da NAD+ in acido piruvico e poi convertito in G6P attraverso la via gluconeogenica. L’accumulo di G6P inibisce la conversione del lattato in piruvato. Il livello di acido lattico aumenta durante il digiuno mentre il glucosio diminuisce. Nelle persone con GSD I, può non scendere completamente alla normalità anche quando vengono ripristinati i normali livelli di glucosio.
L’ipertrigliceridemia derivante da una produzione amplificata di trigliceridi è un altro effetto indiretto della gluconeogenesi alterata, amplificata da livelli di insulina cronicamente bassi. Durante il digiuno, la normale conversione dei trigliceridi in acidi grassi liberi, chetoni e infine acetil-CoA è compromessa. I livelli di trigliceridi nella GSD I possono raggiungere diverse volte la norma e servono come indice clinico di “controllo metabolico”.
L’iperuricemia deriva da una combinazione di aumento della generazione e diminuzione dell’escrezione di acido urico, che viene generato quando vengono metabolizzate maggiori quantità di G6P attraverso la via del pentoso fosfato. È anche un sottoprodotto della degradazione delle purine. L’acido urico compete con l’acido lattico e altri acidi organici per l’escrezione renale nelle urine. Nella GSD I l’aumento della disponibilità di G6P per la via del pentoso fosfato, l’aumento del tasso di catabolismo e la diminuzione dell’escrezione urinaria a causa degli alti livelli di acido lattico si combinano per produrre livelli di acido urico diverse volte superiori al normale. Anche se l’iperuricemia è asintomatica per anni, i danni ai reni e alle articolazioni maturano gradualmente.
Lattato elevato e acidosi latticaModifica
Livelli elevati di acido lattico nel sangue sono osservati in tutte le persone con GSD I, a causa della gluconeogenesi alterata. Gli aumenti basali generalmente vanno da 4 a 10 mol/mL, che non causano alcun impatto clinico. Tuttavia, durante e dopo un episodio di bassa glicemia, i livelli di lattato aumentano bruscamente fino a superare i 15 mol/mL, la soglia dell’acidosi lattica. I sintomi dell’acidosi lattica includono il vomito e l’iperpnea, entrambi i quali possono esacerbare l’ipoglicemia nell’impostazione di GSD I. Nei casi di acidosi lattica acuta, i pazienti hanno bisogno di cure di emergenza per stabilizzare l’ossigeno nel sangue e ripristinare la glicemia. La corretta identificazione dell’acidosi lattica nei bambini non diagnosticati rappresenta una sfida, poiché i primi sintomi sono tipicamente il vomito e la disidratazione, entrambi i quali imitano le infezioni infantili come la gastroenterite o la polmonite. Inoltre, entrambe queste infezioni comuni possono precipitare un’ipoglicemia più grave nei bambini non diagnosticati, rendendo difficile la diagnosi della causa sottostante.
Quando il lattato elevato persiste, l’acido urico, i chetoacidi e gli acidi grassi liberi aumentano ulteriormente il gap anionico. Negli adulti e nei bambini, le alte concentrazioni di lattato causano un disagio significativo nei muscoli. Questo disagio è una forma amplificata della sensazione di bruciore che un corridore può sentire nei quadricipiti dopo lo sprint, che è causato da un breve accumulo di acido lattico. Un adeguato controllo dell’ipoglicemia nella GSD I elimina completamente la possibilità di acidosi lattica.
Urati elevati e complicazioniModifica
Livelli elevati di acido urico si presentano spesso come conseguenza dell’acido lattico elevato nei pazienti con GSD I. Quando i livelli di lattato sono elevati, l’acido lattico presente nel sangue compete per lo stesso meccanismo di trasporto tubulare renale dell’urato, limitando la velocità con cui l’urato può essere eliminato dai reni nelle urine. Se presente, l’aumento del catabolismo delle purine è un ulteriore fattore che contribuisce. Livelli di acido urico da 6 a 12 mg/dl (da 530 a 1060 umol/L) sono comuni tra i pazienti con GSD I, se la malattia non è adeguatamente trattata. In alcune persone colpite, l’uso del farmaco allopurinolo è necessario per abbassare i livelli di urato nel sangue. Le conseguenze dell’iperuricemia tra i pazienti con GSD I includono lo sviluppo di calcoli renali e l’accumulo di cristalli di acido urico nelle articolazioni, con conseguente malattia renale e gotta, rispettivamente.
Iperlipidemia ed effetti plasmaticiModifica
I trigliceridi elevati nella GSD I risultano da una bassa insulina sierica nei pazienti con frequenti ipoglicemie prolungate. Può anche essere causato da accumulo intracellulare di glucosio-6-fosfato con derivazione secondaria in piruvato, che viene convertito in Acetil-CoA, che viene trasportato al citosol dove avviene la sintesi di acidi grassi e colesterolo. I trigliceridi al di sopra dell’intervallo di 3,4 mmol/L (300 mg/dL) possono produrre una lipemia visibile e anche una lieve pseudoiponatriemia dovuta a una ridotta frazione acquosa del plasma sanguigno. Nella GSD I, il colesterolo è tipicamente solo leggermente elevato rispetto ad altri lipidi.
EpatomegaliaModifica
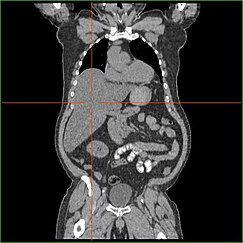
La compromissione della capacità del fegato di effettuare la gluconeogenesi porta all’epatomegalia clinicamente evidente. Senza questo processo, il corpo non è in grado di liberare il glicogeno dal fegato e convertirlo in glucosio nel sangue, portando ad un accumulo di glicogeno immagazzinato nel fegato. L’epatomegalia dovuta all’accumulo di glicogeno immagazzinato nel fegato è considerata una forma di malattia del fegato grasso non alcolica. I pazienti con GSD I presentano un certo grado di epatomegalia per tutta la vita, ma la gravità è spesso legata al consumo di carboidrati alimentari in eccesso. Sono possibili riduzioni della massa del fegato, poiché la maggior parte dei pazienti conserva una funzione epatica residua che permette la liberazione del glicogeno immagazzinato ad un ritmo limitato.
I pazienti con GSD I presentano spesso un’epatomegalia dal momento della nascita. Nello sviluppo fetale, il glucosio materno trasferito al feto previene l’ipoglicemia, ma lo stoccaggio del glucosio come glicogeno nel fegato porta all’epatomegalia. Non ci sono prove che questa epatomegalia presenti alcun rischio per il corretto sviluppo fetale.
L’epatomegalia nella GSD di tipo I si verifica generalmente senza ingrossamento simpatico della milza. I pazienti GSD Ib possono presentare splenomegalia, ma questo è legato all’uso di filgrastim per trattare la neutropenia in questo sottotipo, non l’epatomegalia in comorbilità. L’epatomegalia persisterà in una certa misura per tutta la vita, spesso causando la sporgenza dell’addome, e nei casi gravi può essere palpabile all’altezza dell’ombelico o sotto di esso. Nella malattia del fegato grasso non alcolica legata alla GSD, la funzione epatica è di solito risparmiata, con gli enzimi epatici e la bilirubina che rimangono nella norma. Tuttavia, la funzione epatica può essere influenzata da altre complicazioni epatiche in età adulta, compreso lo sviluppo di adenomi epatici.
Adenomi epaticiModifica
L’eziologia specifica degli adenomi epatici nella GSD I rimane sconosciuta, nonostante la ricerca in corso. Il tipico paziente con GSD I che presenta almeno un adenoma è un adulto, anche se le lesioni sono state osservate in pazienti giovani come quattordici anni. Gli adenomi, composti da neoplasie eterogenee, possono presentarsi singolarmente o in multipli. Le stime sul tasso di conversione di un adenoma epatocellulare in carcinoma epatocellulare nella GSD I vanno dallo 0% all’11%, e quest’ultima cifra rappresenta la ricerca più recente. Una ragione per l’aumento delle stime è la crescente popolazione di pazienti con GSD I che sopravvivono fino all’età adulta, quando si sviluppa la maggior parte degli adenomi.
Le norme di trattamento prevedono l’osservazione regolare del fegato tramite risonanza magnetica o TAC per monitorare le anomalie strutturali. Gli adenomi epatici possono essere erroneamente identificati come iperplasia nodulare focale nella diagnostica per immagini, anche se questa condizione è rara. Tuttavia, gli adenomi epatici in GSD I coinvolgono unicamente la deposizione ialina diffusa di Mallory, che è altrimenti comunemente osservata nell’iperplasia nodulare focale. A differenza dei comuni adenomi epatici legati alla contraccezione orale, l’emorragia nei pazienti con GSD I è rara.
Mentre la ragione dell’alta prevalenza di adenomi nella GSD I non è chiara, la ricerca fin dagli anni ’70 ha implicato il glucagone sierico come potenziale driver. Negli studi, i pazienti che sono stati messi su un regime dietetico per mantenere lo zucchero nel sangue in un range normale che va da 72 a 108 mg/dL (4.0 a 6.0 mmol/L) hanno mostrato una minore probabilità di sviluppare adenomi. Inoltre, i pazienti con glicemia ben controllata hanno costantemente visto una riduzione delle dimensioni e del numero di adenomi epatici, suggerendo che gli adenomi possono essere causati da squilibri di agenti epatotropi come insulina sierica e soprattutto glucagone sierico nel fegato.
Ipertensione portaleModifica
OsteopeniaModifica
I pazienti con GSD I svilupperanno spesso osteopenia. L’eziologia specifica della bassa densità minerale ossea nella GSD non è nota, anche se è fortemente associata con lo scarso controllo metabolico. L’osteopenia può essere causata direttamente dall’ipoglicemia o dalle conseguenze endocrine e metaboliche che ne derivano. I miglioramenti nel controllo metabolico hanno costantemente dimostrato di prevenire o invertire l’osteopenia clinicamente rilevante nei pazienti con GSD I. Nei casi in cui l’osteopenia progredisce con l’età, la densità minerale ossea nelle costole è tipicamente più grave che nelle vertebre. In alcuni casi il T-score della densità minerale ossea scenderà al di sotto di -2,5, indicando l’osteoporosi. C’è una certa evidenza che l’osteopenia può essere collegata alle anomalie renali associate nella GSD I, in particolare l’iperfiltrazione glomerulare. La condizione sembra anche rispondere alla supplementazione di calcio. In molti casi la densità minerale ossea può aumentare e tornare all’intervallo normale dato un adeguato controllo metabolico e la supplementazione di calcio da sola, invertendo l’osteopenia.
Effetti renaliModifica
I reni sono di solito dal 10 al 20% ingranditi con glicogeno immagazzinato. Negli adulti con GSD I, il danno glomerulare cronico simile alla nefropatia diabetica può portare all’insufficienza renale. La GSD I può presentarsi con varie complicazioni renali. Anomalie tubulari renali legate all’iperlattatemia si vedono presto nella vita, probabilmente perché l’acidosi lattica prolungata è più probabile che si verifichi nell’infanzia. Ciò presenterà spesso come sindrome di Fanconi con derangements multipli del riassorbimento tubolare renale, compreso acidosi tubulare con spreco del bicarbonato e del fosfato. Queste anomalie tubulari nella GSD I sono tipicamente rilevate e monitorate dal calcio urinario. A lungo termine queste alterazioni possono esacerbare la nefropatia da acido urico, altrimenti guidata dall’iperlattatemia. Nell’adolescenza e oltre, la malattia glomerulare può svilupparsi indipendentemente, presentandosi inizialmente come iperfiltrazione glomerulare indicata da un elevato eGFR urinario.
SplenomegaliaModifica
L’ingrandimento della milza (splenomegalia) è comune nella GSD I e ha due cause principali. Nella GSD Ia, la splenomegalia può essere causata da una relazione tra il fegato e la milza che fa crescere o rimpicciolire l’uno o l’altro per adattarsi alle dimensioni relative dell’altro, in misura ridotta. Nella GSD Ib, è un effetto collaterale dell’uso di filgrastim per trattare la neutropenia.
Effetti intestinaliModifica
Il coinvolgimento intestinale può causare un lieve malassorbimento con feci grasse (steatorrea), ma di solito non richiede alcun trattamento.
Rischio di infezioneModifica
La neutropenia è una caratteristica distintiva della GSD Ib, assente nella GSD Ia. La causa microbiologica della neutropenia nella GSD Ib non è ben compresa. In generale, il problema deriva da un metabolismo cellulare compromesso nei neutrofili, con conseguente apoptosi accelerata dei neutrofili. La neutropenia nella GSD è caratterizzata sia da una diminuzione della conta assoluta dei neutrofili che da una diminuzione della funzione dei neutrofili. I neutrofili utilizzano una specifica via metabolica G6P che si basa sulla presenza di G6Pase-β o G6PT per mantenere l’omeostasi energetica all’interno della cellula. L’assenza di G6PT nella GSD Ib limita questo percorso, portando allo stress del reticolo endoplasmatico, allo stress ossidativo all’interno del neutrofilo, innescando l’apoptosi prematura. Il fattore stimolante le colonie di granulociti (G-CSF), disponibile come filgrastim, può ridurre il rischio di infezione. In alcuni casi, G-CSF formulato come pegfilgrastim, venduto con il nome commerciale Neulasta, può essere utilizzato come alternativa ad azione lenta, che richiede un dosaggio meno frequente.
Trombocitopenia e problemi di coagulazione del sangueModifica
L’aggregazione piastrinica alterata è una conseguenza non comune di ipoglicemia cronica, vista nei pazienti con GSD I. La ricerca ha dimostrato una diminuzione della funzione piastrinica, caratterizzata da una diminuzione del consumo di protrombina, reazioni di aggregazione anormali, tempo di sanguinamento prolungato e bassa adesività piastrinica. La gravità della disfunzione piastrinica è tipicamente correlata alle condizioni cliniche, con i casi più gravi correlati all’acidosi lattica e alla grave lipidemia. Può causare emorragie clinicamente significative, soprattutto epistassi. Inoltre, i pazienti con GSD I possono presentare trombocitopenia come conseguenza della splenomegalia. Nell’impostazione della splenomegalia vari fattori ematologici possono essere sequestrati nei tessuti della milza mentre il sangue viene filtrato attraverso l’organo. Questo può diminuire i livelli di piastrine disponibili nel flusso sanguigno, portando alla trombocitopenia.
Effetti sullo sviluppoModifica
Il ritardo dello sviluppo è un potenziale effetto secondario dell’ipoglicemia cronica o ricorrente, ma è almeno teoricamente prevenibile. Le normali cellule neuronali e muscolari non esprimono la glucosio-6-fosfatasi, e quindi non sono influenzate direttamente dalla GSD I. Tuttavia, senza un adeguato trattamento dell’ipoglicemia, il fallimento della crescita deriva comunemente da livelli di insulina cronicamente bassi, acidosi persistente, elevazione cronica degli ormoni catabolici e insufficienza calorica (o malassorbimento). I ritardi di sviluppo più drammatici sono spesso la causa di gravi (non solo persistenti) episodi di ipoglicemia.