- Normal kolhydratbalans och upprätthållande av blodglukosnivåerRedigera
- PatofysiologiRedigera
- Förhöjt laktat och mjölkacidosRedigera
- Förhöjt urat och komplikationerRedigera
- Hyperlipidemi och plasmaeffekterRedigera
- HepatomegaliRedigera
- LeveradenomEdit
- Portal hypertensionRedigera
- OsteopeniEdit
- NjurpåverkanRedigera
- SplenomegaliRedigera
- TarmeffekterEdit
- InfektionsriskEdit
- Trombocytopeni och problem med blodkoagulationRedigera
- UtvecklingseffekterRedigera
Normal kolhydratbalans och upprätthållande av blodglukosnivåerRedigera
Glykogen i levern och (i mindre utsträckning) i njurarna fungerar som en form av lagrat, snabbt tillgängligt glukos, så att blodglukosnivån kan upprätthållas mellan måltiderna. Under cirka tre timmar efter en kolhydrathaltig måltid styr höga insulinnivåer levercellerna att ta glukos från blodet, omvandla det till glukos-6-fosfat (G6P) med enzymet glukokinas och lägga till G6P-molekylerna i ändarna av glykogenkedjorna (glykogensyntes). Överskott av G6P shuntas också in i produktionen av triglycerider och exporteras för lagring i fettvävnad som fett.
När matsmältningen av en måltid är avslutad sjunker insulinnivåerna och enzymsystemen i levercellerna börjar avlägsna glukosmolekyler från glykogensträngar i form av G6P. Denna process kallas glykogenolys. G6P stannar kvar i levercellen om inte fosfatet klyvs av glukos-6-fosfatas. Denna avfosforyleringsreaktion ger upphov till fri glukos och fria PO
4-anjoner. De fria glukosmolekylerna kan transporteras ut ur levercellerna till blodet för att upprätthålla en tillräcklig tillförsel av glukos till hjärnan och andra organ i kroppen. Glykogenolysen kan tillgodose glukosbehovet hos en vuxen kropp i 12-18 timmar.
När fastan fortsätter i mer än några timmar tillåter fallande insulinnivåer katabolism av muskelprotein och triglycerider från fettvävnad. Produkterna från dessa processer är aminosyror (främst alanin), fria fettsyror och mjölksyra. Fria fettsyror från triglycerider omvandlas till ketoner och till acetyl-CoA. Aminosyror och mjölksyra används för att syntetisera nytt G6P i leverceller genom glukoneogenesen. Det sista steget i normal glukoneogenes, liksom det sista steget i glykogenolysen, är defosforylering av G6P av glukos-6-fosfatas till fri glukos och PO
Därmed medierar glukos-6-fosfatas det sista, viktiga steget i båda de två huvudprocesserna för glukosproduktion under fastan. Effekten förstärks eftersom de resulterande höga nivåerna av glukos-6-fosfat hämmar tidigare nyckelsteg i både glykogenolysen och glukoneogenesen.
PatofysiologiRedigera
De huvudsakliga metaboliska effekterna av brist på glukos-6-fosfatas är hypoglykemi, mjölksyraacidos, hypertriglyceridemi och hyperurikemi.

Hypoglykemin vid GSD I benämns som ”fastande”, eller ”postabsorptiv”, vanligen cirka 4 timmar efter fullständig matsmältning av en måltid. Denna oförmåga att upprätthålla adekvata blodglukosnivåer under fastan beror på den kombinerade försämringen av både glykogenolysen och glukoneogenesen. Fastandehypoglykemi är ofta det största problemet hos GSD I, och vanligtvis det problem som leder till diagnosen. Kronisk hypoglykemi ger upphov till sekundära metaboliska anpassningar, inklusive kroniskt låga insulinnivåer och höga nivåer av glukagon och kortisol.
Mjölksacidos uppstår på grund av försämrad glukoneogenes. Mjölksyra bildas både i levern och musklerna och oxideras av NAD+ till pyruvsyra och omvandlas sedan via den glukoneogena vägen till G6P. Ackumulering av G6P hämmar omvandlingen av laktat till pyruvat. Mjölksyranivån stiger under fasta när glukoshalten sjunker. Hos personer med GSD I kan det hända att den inte sjunker helt till det normala även när normala glukosnivåer återställs.
Hypertriglyceridemi till följd av förstärkt triglyceridproduktion är en annan indirekt effekt av nedsatt glukoneogenes, som förstärks av kroniskt låga insulinnivåer. Under fasta är den normala omvandlingen av triglycerider till fria fettsyror, ketoner och slutligen acetyl-CoA nedsatt. Triglyceridnivåerna hos GSD I kan uppgå till flera gånger det normala och fungerar som ett kliniskt index för ”metabolisk kontroll”.
Hyperurikemi beror på en kombination av ökad bildning och minskad utsöndring av urinsyra, som bildas när ökade mängder G6P metaboliseras via pentosfosfatvägen. Det är också en biprodukt vid nedbrytning av puriner. Urinsyra konkurrerar med mjölksyra och andra organiska syror om renal utsöndring i urinen. Vid GSD I leder ökad tillgång till G6P för pentosfosfatvägen, ökad katabolism och minskad urinutsöndring på grund av höga nivåer av mjölksyra tillsammans till flera gånger högre urinsyrenivåer än normalt. Även om hyperurikemi är asymtomatisk i åratal, så ackumuleras gradvis njur- och ledskador.
Förhöjt laktat och mjölkacidosRedigera
Höga nivåer av mjölksyra i blodet observeras hos alla personer med GSD I, på grund av nedsatt glukoneogenes. Förhöjningar vid baslinjen varierar i allmänhet mellan 4 och 10 mol/mL, vilket inte orsakar någon klinisk påverkan. Under och efter en episod av lågt blodsocker kommer dock laktatnivåerna att plötsligt stiga till över 15 mol/mL, vilket är tröskeln för mjölkacidos. Symtom på laktacidos är bland annat kräkningar och hyperpné, som båda kan förvärra hypoglykemin vid GSD I. Vid akut laktacidos behöver patienterna akut vård för att stabilisera syrehalten i blodet och återställa blodglukos. Korrekt identifiering av mjölkacidos hos odiagnostiserade barn är en utmaning, eftersom de första symptomen vanligtvis är kräkningar och uttorkning, som båda efterliknar barninfektioner som gastroenterit eller lunginflammation. Dessutom kan båda dessa vanliga infektioner påskynda allvarligare hypoglykemi hos odiagnostiserade barn, vilket gör det svårt att diagnostisera den underliggande orsaken.
När förhöjt laktat kvarstår ökar urinsyra, ketosyror och fria fettsyror anjongapet ytterligare. Hos vuxna och barn orsakar de höga koncentrationerna av laktat betydande obehag i musklerna. Detta obehag är en förstärkt form av den brännande känsla som en löpare kan känna i quadriceps efter en sprint, vilket orsakas av en kortvarig uppbyggnad av mjölksyra. Korrekt kontroll av hypoglykemi hos GSD I eliminerar helt och hållet möjligheten till mjölksyraacidos.
Förhöjt urat och komplikationerRedigera
Höga nivåer av urinsyra uppträder ofta som en följd av förhöjd mjölksyra hos GSD I-patienter. När laktatnivåerna är förhöjda konkurrerar blodburen mjölksyra med urat om samma transportmekanism i njurtuberna som urat, vilket begränsar hastigheten med vilken urat kan rensas bort av njurarna till urinen. Om det finns en ökad purinkatabolism är en ytterligare bidragande faktor. Urinsyrenivåer på 6-12 mg/dl (530-1060 umol/L) är vanliga bland GSD I-patienter, om sjukdomen inte behandlas på rätt sätt. Hos vissa drabbade personer är det nödvändigt att använda läkemedlet allopurinol för att sänka uratnivåerna i blodet. Konsekvenser av hyperurikemi bland GSD I-patienter är bland annat utveckling av njursten och ansamling av urinsyrakristaller i lederna, vilket leder till njursjukdom respektive gikt.
Hyperlipidemi och plasmaeffekterRedigera
Höjda triglycerider hos GSD I är ett resultat av lågt seruminsulin hos patienter med frekvent långvarig hypoglykemi. Den kan också orsakas av intracellulär ackumulering av glukos-6-fosfat med sekundär shuntning till pyruvat, som omvandlas till acetyl-CoA, som transporteras till cytosolen där syntesen av fettsyror och kolesterol sker. Triglycerider över 3,4 mmol/L (300 mg/dL) kan ge synlig lipemi och till och med en mild pseudohyponatremi på grund av en minskad vattenfraktion i blodplasman. Vid GSD I är kolesterolet vanligtvis endast lätt förhöjt jämfört med andra lipider.
HepatomegaliRedigera
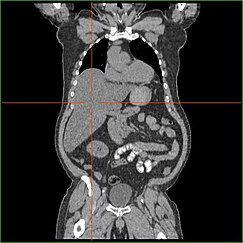
Nedsättning av leverns förmåga att utföra glukoneogenes leder till kliniskt synlig hepatomegali. Utan denna process kan kroppen inte frigöra glykogen från levern och omvandla det till blodglukos, vilket leder till en ansamling av lagrat glykogen i levern. Hepatomegali på grund av ackumulering av lagrat glykogen i levern anses vara en form av icke-alkoholisk fettleversjukdom. GSD I-patienter uppvisar en viss grad av hepatomegali under hela livet, men svårighetsgraden hänger ofta samman med konsumtionen av överskott av kolhydrater i kosten. Det är möjligt att minska leverns massa, eftersom de flesta patienter har kvar en kvarvarande leverfunktion som gör det möjligt att frigöra lagrat glykogen i begränsad takt.
GSD I-patienter uppvisar ofta hepatomegali från födseln. Under fosterutvecklingen förhindrar glukos från moderns sida som överförs till fostret hypoglykemi, men lagring av glukos som glykogen i levern leder till hepatomegali. Det finns inga bevis för att denna hepatomegali utgör någon risk för en korrekt fosterutveckling.
Hepatomegali vid GSD typ I uppträder i allmänhet utan sympatisk förstoring av mjälten. GSD Ib-patienter kan uppvisa splenomegali, men detta är kopplat till användningen av filgrastim för att behandla neutropeni i denna subtyp, inte komorbid hepatomegali. Hepatomegali kommer att kvarstå i viss grad under hela livet, vilket ofta leder till att buken sticker ut, och i svåra fall kan den vara palpabel vid eller under naveln. Vid GSD-relaterad icke-alkoholisk fettleversjukdom är leverfunktionen vanligen skonad, med leverenzymer och bilirubin som ligger inom det normala intervallet. Leverfunktionen kan dock påverkas av andra leverkomplikationer i vuxen ålder, inklusive utveckling av leveradenom.
LeveradenomEdit
Den specifika etiologin för leveradenom hos GSD I är fortfarande okänd, trots pågående forskning. Den typiska GSD I-patienten som presenterar minst ett adenom är en vuxen person, även om lesioner har observerats hos patienter så unga som fjorton år. Adenom, som består av heterogena neoplasmer, kan förekomma enskilt eller i flera exemplar. Uppskattningar av hur ofta ett hepatocellulärt adenom omvandlas till hepatocellulärt karcinom i GSD I varierar från 0 % till 11 %, där den senare siffran representerar nyare forskning. En anledning till den ökande uppskattningen är den växande populationen av GSD I-patienter som överlever in i vuxen ålder, då de flesta adenom utvecklas.
Behandlingsnormerna föreskriver regelbunden observation av levern med MRT eller datortomografi för att övervaka strukturella avvikelser. Leveradenom kan felidentifieras som fokal nodulär hyperplasi vid diagnostisk avbildning, även om detta tillstånd är sällsynt. Leverns adenom i GSD I har dock en unik karaktär med diffus Mallory-hyalinavlagring, som annars ofta observeras vid fokal nodulär hyperplasi. Till skillnad från vanliga hepatiska adenom relaterade till orala preventivmedel är blödningar hos GSD I-patienter sällsynta.
Och även om orsaken till den höga prevalensen av adenom i GSD I är oklar, har forskning sedan 1970-talet implicerat serumglukagon som en potentiell drivkraft. I studier har patienter som fått en diet för att hålla blodsockret i ett normalt intervall som sträcker sig från 72 till 108 mg/dL (4,0 till 6,0 mmol/L) visat en minskad sannolikhet för att utveckla adenom. Dessutom har patienter med välkontrollerat blodsocker konsekvent sett en minskning av storleken och antalet leveradenom, vilket tyder på att adenom kan orsakas av obalanser av hepatotropa ämnen som seruminsulin och särskilt serumglukagon i levern.
Portal hypertensionRedigera
OsteopeniEdit
Patienter med GSD I utvecklar ofta osteopeni. Den specifika etiologin till låg benmineraltäthet hos GSD är inte känd, även om den är starkt förknippad med dålig metabolisk kontroll. Osteopeni kan vara direkt orsakad av hypoglykemi eller de resulterande endokrina och metaboliska följderna. Förbättrad metabolisk kontroll har konsekvent visat sig förhindra eller vända kliniskt relevant osteopeni hos GSD I-patienter. I de fall där osteopeni utvecklas med åldern är benmineraltätheten i revbenen vanligtvis allvarligare än i kotorna. I vissa fall sjunker benmineraltäthetens T-score under -2,5, vilket tyder på osteoporos. Det finns vissa bevis för att osteopeni kan vara kopplad till associerade njuravvikelser hos GSD I, särskilt glomulär hyperfiltrering. Tillståndet verkar också reagera på kalciumtillskott. I många fall kan benmineraltätheten öka och återgå till det normala intervallet givet korrekt metabolisk kontroll och enbart kalciumtillskott, vilket reverserar osteopeni.
NjurpåverkanRedigera
Njurarna är vanligen 10 till 20 % förstorade med lagrat glykogen. Hos vuxna med GSD I kan kronisk glomerulär skada som liknar diabetisk nefropati leda till njursvikt. GSD I kan uppvisa olika njurkomplikationer. Renala tubulära abnormiteter relaterade till hyperlaktatemi ses tidigt i livet, troligen på grund av att långvarig laktacidos är mer sannolikt att inträffa i barndomen. Detta kommer ofta att visa sig som Fanconis syndrom med flera störningar i den renala tubulära reabsorptionen, inklusive tubulär acidos med bikarbonat- och fosfatförlust. Dessa tubulära avvikelser hos GSD I upptäcks och övervakas vanligtvis med hjälp av kalcium i urinen. På lång sikt kan dessa störningar förvärra urinsyranefropati, som annars drivs av hyperlaktatemi. I tonåren och därefter kan en glomerulär sjukdom utvecklas oberoende av varandra, som inledningsvis presenteras som glomerulär hyperfiltrering som indikeras av ett förhöjt eGFR i urinen.
SplenomegaliRedigera
En utvidgning av mjälten (splenomegali) är vanlig hos GSD I och har två primära orsaker. Hos GSD Ia kan splenomegali orsakas av ett förhållande mellan levern och mjälten som gör att endera växer eller krymper för att matcha den andra partens relativa storlek, i minskad grad. Hos GSD Ib är det en biverkning av användningen av filgrastim för att behandla neutropeni.
TarmeffekterEdit
Intestinal inblandning kan orsaka mild malabsorption med fet avföring (steatorré), men kräver vanligen ingen behandling.
InfektionsriskEdit
Neutropeni är ett utmärkande kännetecken för GSD Ib, saknas hos GSD Ia. Den mikrobiologiska orsaken till neutropeni vid GSD Ib är inte väl klarlagd. I stort sett uppstår problemet på grund av komprometterad cellulär metabolism i neutrofilen, vilket resulterar i accelererad neutrofil apoptos. Neutropeni vid GSD kännetecknas av både en minskning av det absoluta antalet neutrofiler och en försämrad neutrofil funktion. Neutrofiler använder en specifik G6P-metabolisk väg som är beroende av närvaron av G6Pase-β eller G6PT för att upprätthålla energihomeostasen i cellen. Avsaknaden av G6PT i GSD Ib begränsar denna väg, vilket leder till stress i det endoplasmatiska retikulumet, oxidativ stress i neutrofilen och utlöser för tidig apoptos. Granulocytkolonistimulerande faktor (G-CSF), som finns som filgrastim, kan minska risken för infektion. I vissa fall kan G-CSF formulerat som pegfilgrastim, som säljs under handelsnamnet Neulasta, användas som ett långsamt verkande alternativ som kräver mindre frekvent dosering.
Trombocytopeni och problem med blodkoagulationRedigera
Svårad trombocytaggregation är en ovanlig konsekvens av kronisk hypoglykemi som ses hos GSD I-patienter. Forskning har visat på nedsatt trombocytfunktion, som kännetecknas av minskad protrombinförbrukning, onormala aggregeringsreaktioner, förlängd blödningstid och låg trombocytadhäsivitet. Svårighetsgraden av trombocytdysfunktion korrelerar vanligtvis med det kliniska tillståndet, där de allvarligaste fallen korrelerar med laktacidos och allvarlig lipidemi. Det kan orsaka kliniskt betydande blödningar, särskilt epistaxis. Dessutom kan GSD I-patienter uppvisa trombocytopeni till följd av splenomegali. I samband med splenomegali kan olika hematologiska faktorer sekquerteras i mjältens vävnader när blodet filtreras genom organet. Detta kan minska nivåerna av trombocyter som är tillgängliga i blodomloppet, vilket leder till trombocytopeni.
UtvecklingseffekterRedigera
Utvecklingsförsening är en potentiell sekundär effekt av kronisk eller återkommande hypoglykemi, men är åtminstone teoretiskt möjlig att förebygga. Normala neuronal- och muskelceller uttrycker inte glukos-6-fosfatas och påverkas därför inte direkt av GSD I. Om hypoglykemin inte behandlas på rätt sätt resulterar tillväxtsvårigheter vanligen i kroniskt låga insulinnivåer, ihållande acidos, kronisk förhöjning av kataboliska hormoner och kaloriinsufficiens (eller malabsorption). De mest dramatiska utvecklingsförseningarna är ofta orsaken till allvarliga (inte bara ihållande) episoder av hypoglykemi.