Une fille de 8 ans a été adressée par l’endocrinologie au service de dermatologie pour l’évaluation de lésions légèrement prurigineuses dans les deux aisselles. Les lésions s’étaient développées sur une longue période et étaient plus sévères dans l’aisselle gauche. La patiente, dont les parents étaient originaires de Roumanie, était suivie par l’endocrinologie pédiatrique en raison d’une puberté précoce, avec un taux de testostérone légèrement élevé (test de Synacthen normal). Elle avait également présenté un épisode de gastro-entérite aiguë avec insuffisance hépatique, qui a nécessité un suivi pendant 3 ans.
L’examen physique a révélé des papules élastiques jaunâtres mesurant entre 1 et 2 mm, avec une distribution périfolliculaire, regroupées en plaques localisées dans les deux aisselles, avec un regroupement plus disparate du côté gauche (figures 1 et 2).
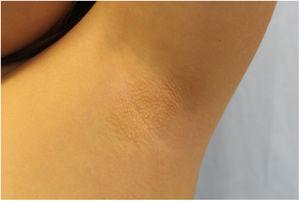
Papules périfolliculaires confluentes de taille millimétrique, de couleur jaunâtre et de consistance élastique, au niveau de l’aisselle gauche.
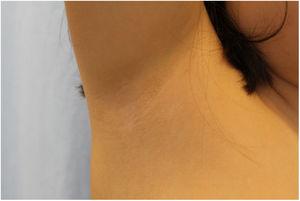
Lésions moins sévères dans l’aisselle droite.
L’histologie d’une biopsie à l’emporte-pièce d’une des lésions a révélé une dilatation de l’infundibulum folliculaire et une agrégation périfolliculaire de cellules spumeuses (figure 3). Ces résultats étaient compatibles avec un diagnostic de maladie de Fox-Fordyce (FFD).
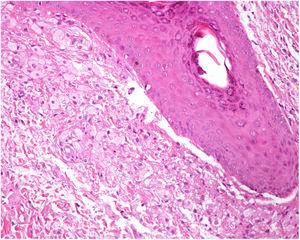
Accumulation de cellules spumeuses adjacentes à un follicule pileux.
L’évolution clinique a été favorable, avec une amélioration spontanée des lésions. Les seuls antécédents familiaux pertinents étaient que sa mère avait signalé une hyperhidrose et une bromhidrose axillaire, et elle est donc sortie de l’hôpital avec un antisudorifique local à la demande (chlorhydrate d’aluminium, 15%). Il n’a pas été possible d’établir une relation temporelle entre la maladie endocrinologique sous-jacente de la patiente et l’apparition ou l’amélioration des lésions.
La FFD, également appelée miliaire apocrine, a été décrite pour la première fois en 1902 par George Henry Fox et John Addison Fordyce. C’est une maladie chronique rare qui apparaît typiquement chez les femmes postpubères (15-35 ans).1 La présentation dans l’enfance est exceptionnelle.2 L’étiologie est inconnue et la cause est probablement multifactorielle.3 Son association avec des facteurs hormonaux a été postulée, étant donné la tranche d’âge dans laquelle elle apparaît et la rémission fréquente de la maladie pendant la grossesse et la ménopause, bien qu’il n’ait pas été possible de mettre en évidence une quelconque anomalie hormonale chez les patients.Nous ne savons donc pas si une association existe avec la puberté précoce de notre patiente. Nous n’avons pas trouvé d’association antérieure avec l’insuffisance hépatique dans la littérature.
L’apparition de la maladie comme effet indésirable des techniques d’épilation au laser ou à la lumière pulsée intense (IPL) a été rapportée.4 Dans ce cas, les résultats cliniques et histologiques sont très similaires à ceux de la FFD primaire.
Elle se présente cliniquement comme de petites papules périfolliculaires prurigineuses en forme de dôme de la couleur de la peau jaunâtre-saine. La localisation la plus fréquente est les aisselles mais les lésions peuvent également se présenter dans la région périaréolaire ou anogénitale et, plus rarement, sur les cuisses, la région périombilicale et le sternum. La maladie peut être associée à une perte de cheveux dans les zones affectées et à une hypohidrose.
Les lésions s’améliorent avec la grossesse et l’utilisation de contraceptifs hormonaux, et s’aggravent avec la chaleur, l’eau chaude, le stress et le cycle menstruel.1
La présentation clinique de la maladie est constante et similaire. La formation d’un bouchon de kératine dans l’infundibulum folliculaire, qui obstrue également le canal apocrine, peut être le déclencheur.3 La caractéristique la plus constante est la dilatation de l’infundibulum et l’hyperkératose de l’épithélium infundibulaire.5 La découverte la plus spécifique est la xanthomatose périfolliculaire.6
On pensait traditionnellement que cette maladie n’impliquait que les glandes apocrines. Cependant, des résultats histologiques récents suggèrent que des variantes eccrines et non folliculaires (impliquant les glandes apocrines) peuvent exister.3
Le diagnostic différentiel clinique de cette entité devrait fondamentalement inclure le syringome éruptif, le lichen nitidus, le trichostasis spinulosa, le syndrome de Graham-Little-Piccardi-Lasseur et la maladie de Darier.1,7,8
Comme il s’agit d’une maladie rare, aucune étude comparative n’est disponible et le traitement reste flou. De multiples traitements ont été décrits, tous avec des résultats variables. Les corticostéroïdes topiques ou intralésionnels sont considérés comme le traitement de choix. Les rétinoïdes topiques et systémiques, les antibiotiques topiques tels que la clindamycine, les antisudorifiques, les contraceptifs oraux et les inhibiteurs topiques de la calcineurine ont également été utilisés. Le curetage et l’électrocoagulation, les techniques de liposuccion modifiées et, plus récemment, le laser CO2 fractionné, les micro-ondes et la toxine botulique A ont été utilisés dans les cas réfractaires.9-11
La rémission spontanée, comme dans le cas de notre patient, est anecdotique.
En résumé, nous rapportons un cas de FFD exceptionnel en raison de son apparition à l’âge prépubertaire et de son évolution atypique, avec une résolution spontanée des lésions. Nous ne pouvons pas affirmer la relation de cause à effet avec les anomalies endocrinologiques de la patiente, bien qu’elles jouent probablement un rôle majeur dans le développement de la maladie.
Conflits d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts.