 |
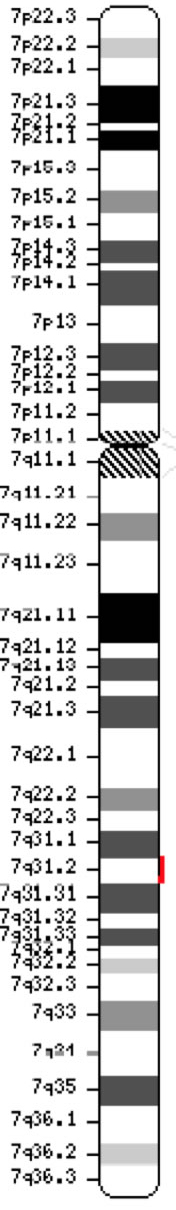
Gen pro regulátor transmembránového vedení cystické fibrózy (CFTR, OMIM #602421) je členem podrodiny C (ABCC7) vázající kazety ATP. Gen, objevený v roce 1989 (Kerem et al, 1989; Riordan et al, 1989; Rommens et al, 1989), se rozkládá na 188 702 bp na dlouhém raménku chromozomu 7 (pás 7q31-q32) (pozice 117 119 358-117 308 719) a skládá se z 27 exonů původně číslovaných 1-24 s dělením a a b pro exony 6, 14 a 17 (Zielenski et al., 1991). Zralý transkript je dlouhý 6 129 nukleotidů včetně otevřeného čtecího rámce o délce 4 440 kódujících bází (referenční sekvence: NM_000492). Ensembl ID genu: ENSG00000001626 Chromosom 7 ( NCBI nebo HGNC) |
Nomenklatura
UmD-CFTR používá doporučené pokyny pro nomenklaturu navržené Human Genome Variation Society (www.hgvs.org/mutnomen/), např, číslování založené na cDNA s A translačního iniciačního kodonu ATG na +1 a exony číslovanými 1 až 27.
U UMD-CFTR je kódovací referenční sekvence NM_000492 s výjimkou c.1408A místo G a referenční proteinová sekvence NP_000483 s výjimkou p.Met470 místo p.Val470.
Pro pohodlí uživatelů, kteří jsou zvyklí na číslování konsorcia databáze mutací cystické fibrózy (www.genet.sickkids.on.ca/cftr/), jsou k dispozici dva soubory ke stažení:
– korespondenční číslování exonů a nukleotidů: běžné číslování (bíle pro exony a černě pro nukleotidy) a doporučené číslování (červeně). U každého exonu je uveden první a poslední nukleotid. (ke stažení) (Zobrazit/skrýt)
– korespondenční tabulka pro názvy variant. (download)
Kódující sekvence (Show/Hide)
Mutace CFTR
Dosud bylo popsáno téměř 1 700 mutací a variací sekvence CFTR (www.genet.sickkids.on.ca/cftr). Celosvětově nejčastější mutací zodpovědnou za cystickou fibrózu je delece tří bází kódující fenylalaninový zbytek na pozici 508 označovaná jako p.Phe508del, která zhoršuje schopnost proteinu CFTR skládat se v endoplazmatickém retikulu (ER), a tím zvyšuje rychlou degradaci proteinu během zpracování v ER. Představuje přibližně 70 % alel CF.
Heterologická exprese plnohodnotné p.Phe508del-CFTR-cDNA prokázala, že tento mutantní protein získává pouze jádrovou glykosylaci, nikoliv komplexní oligosacharidové řetězce, a proto není transportován na povrch buněk, což je v souladu s pozorováním nepřítomnosti mutantního proteinu v čerstvých epiteliích z tkání pacientů. Kromě jiných škodlivých účinků p.Phe508del narušuje správné skládání CFTR a také konformačně maskuje diacidový výstupní kód tří zbytků v NBD1, který se zdá být nezbytný pro export do ER.
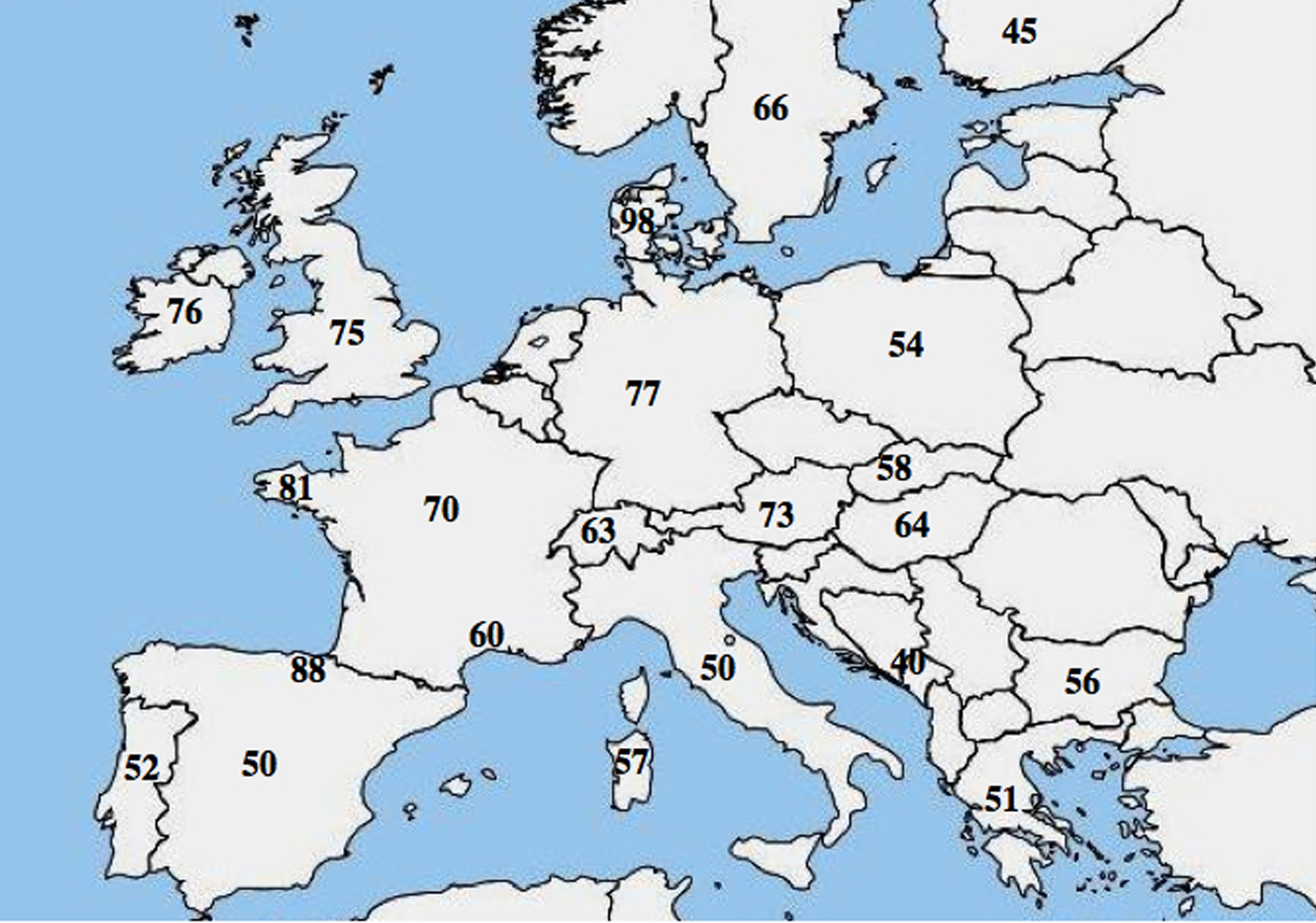
U CF byla zaznamenána molekulární alelická heterogenita; četné studie poukázaly na výrazný rozdíl ve frekvenci p.Phe508del mezi populacemi a ukázaly klesající gradient od severozápadu k jihovýchodu v Evropě.
Třídy mutací CFTR
Mutace v genu CFTR lze rozdělit do šesti tříd podle jejich molekulárních mechanismů a důsledků pro různé aspekty biogeneze, metabolismu a funkce CFTR.
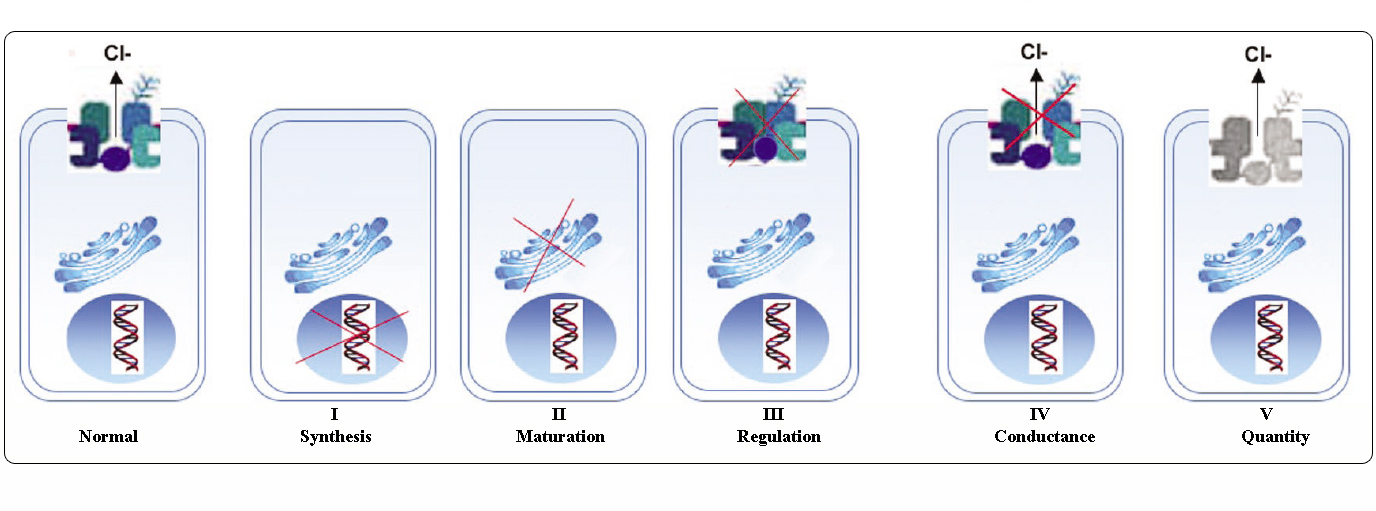
Převzato podle Welshe a Smithe (1993) a upraveno podle Claustrese (RBM online, 2005)
Mutace patřící do tříd I-III a VI propůjčují málo funkční nebo žádný CFTR na apikální membráně a jsou považovány za „těžké“ a obvykle vedou ke klasickému fenotypu CF s pankreatickou insuficiencí (CF-PI), i když závažnost plicního onemocnění může být různá.
Mutace patřící do tříd IV a V zachovávají určitou zbytkovou aktivitu CFTR a propůjčují mírnější fenotyp. U pacientů s alespoň jednou „mírnou“ alelou CF je funkce CFTR obvykle dostatečná pro trávení a plicní onemocnění je méně závažné. U jedinců s mutacemi CFTR, které dávají vznik přibližně 10 % normální hladiny CFTR mRNA, se může vyskytovat pouze jeden příznak, například CBAVD (vrozená oboustranná absence chámovodů) u mužů. U některých jedinců s heterozygotní mutací CFTR může být zvýšené riziko „poruch souvisejících s CFTR“ (CFTR-RD), jako je pankreatitida, sinusitida nebo alergická bronchopulmonální aspergilóza. V těchto případech by mutace CFTR mohly být faktorem genetické predispozice a genetické nebo environmentální modifikátory by mohly hrát aditivní roli.
Seskupení mutací do různých tříd je užitečné pro pochopení mechanismu dysfunkce, nicméně mnoho sekvenčních variací je vzácných/jedinečných a dostupné údaje neumožňují posoudit jejich patogenitu. Navíc jedna mutace může způsobovat více než jeden typ abnormality nebo může propůjčovat variabilní fenotyp.
* Claustres M. 2005. Molekulární patologie lokusu CFTR u mužské neplodnosti. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Komplexnost u monogenního onemocnění. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identifikace genu pro cystickou fibrózu: genetická analýza. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL a další. 1989. Identifikace genu pro cystickou fibrózu: klonování a charakterizace komplementární DNA. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Fyzikální lokalizace dvou DNA markerů úzce spojených s lokusem cystické fibrózy pomocí gelové elektroforézy s pulzním polem. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. Molekulární mechanismy dysfunkce chloridového kanálu CFTR u cystické fibrózy. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Sekvence genomové DNA genu CFTR (cystic fibrosis transmembrane conductance regulator). Genomics 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. Varianta genu CFTR u pacientů s vrozenou absencí vas deferens. Am J Hum Genet 57(4):958-60. PMID: 7573058.
.