 |
O gene regulador de condutância da fibrose cística transmembrana (CFTR, OMIM #602421) é um membro da sub-família ATP-cassete de ligação C (ABCC7). O gene, descoberto em 1989 (Kerem et al, 1989; Riordan et al, 1989; Rommens et al, 1989), abrange 188.702 bp no braço longo do cromossomo 7 (banda 7q31-q32) (posição 117.119.358-117.308.719) e é composto de 27 exons inicialmente numerados 1-24 com subdivisões a e b para os exons 6, 14 e 17 (Zielenski et al., 1991). A transcrição madura tem 6.129 nucleotídeos, incluindo um quadro de leitura aberto de 4.440 bases de codificação (sequência de referência: NM_000492). Ensemblage Gene ID: ENSG00000001626 Cromossoma 7 ( NCBI, ou HGNC) |
Nomenclatura
O UMD-CFTR utiliza as directrizes de nomenclatura recomendadas pela Human Genome Variation Society (www.hgvs.org/mutnomen/), por exemplo, cDNA baseado na numeração com o A do códon de iniciação translacional ATG em +1 e exões numerados de 1 a 27.
No UMD-CFTR a sequência de referência de codificação é NM_000492 com a excepção de c.1408A em vez de G, e a sequência de referência de proteínas é NP_000483 com a excepção de p.Met470 em vez de p.Val470.
Para conveniência dos utilizadores que estão habituados à numeração em consórcio da base de dados de mutações de fibrose cística (www.genet.sickkids.on.ca/cftr/) são fornecidos dois ficheiros para download:
– uma numeração de correspondência para exons e nucleotídeos: numeração comum (em branco para exons e em preto para nucleotídeos) e numeração recomendada (em vermelho). O primeiro e o último nucleotídeos são indicados para cada exão. (download) (Mostrar/Esconder)
– uma tabela de correspondência para os nomes das variações. (download)
Sequência de codificação (Mostrar/Esconder)
As mutações CFTR
Mutações e variações da sequência CFTR foram descritas até agora (www.genet.sickkids.on.ca/cftr). A mutação mais comum no mundo responsável pela fibrose cística é a eliminação de três bases que codificam um resíduo de fenilalanina na posição 508 chamada p.Phe508del, que prejudica a capacidade da proteína CFTR de se dobrar no retículo endoplasmático (ER), aumentando assim a rápida degradação da proteína durante o processamento do ER. Ela é responsável por cerca de 70% dos alelos CF.
Expressão heteróloga de p.Phe508del-CFTR-cDNA de comprimento total demonstrou que esta proteína mutante adquire apenas glicosilação do núcleo e não cadeias complexas de oligossacarídeos e, como tal, não é transportada para a superfície celular, de acordo com a observação da ausência de proteína mutante em epitélios frescos dos tecidos do paciente. Entre outros efeitos prejudiciais, o p.Phe508del prejudica a dobra correta do CFTR e também mascara conformavelmente o código de saída diacídica de três resíduos no NBD1 que parece necessário para a exportação de ER.
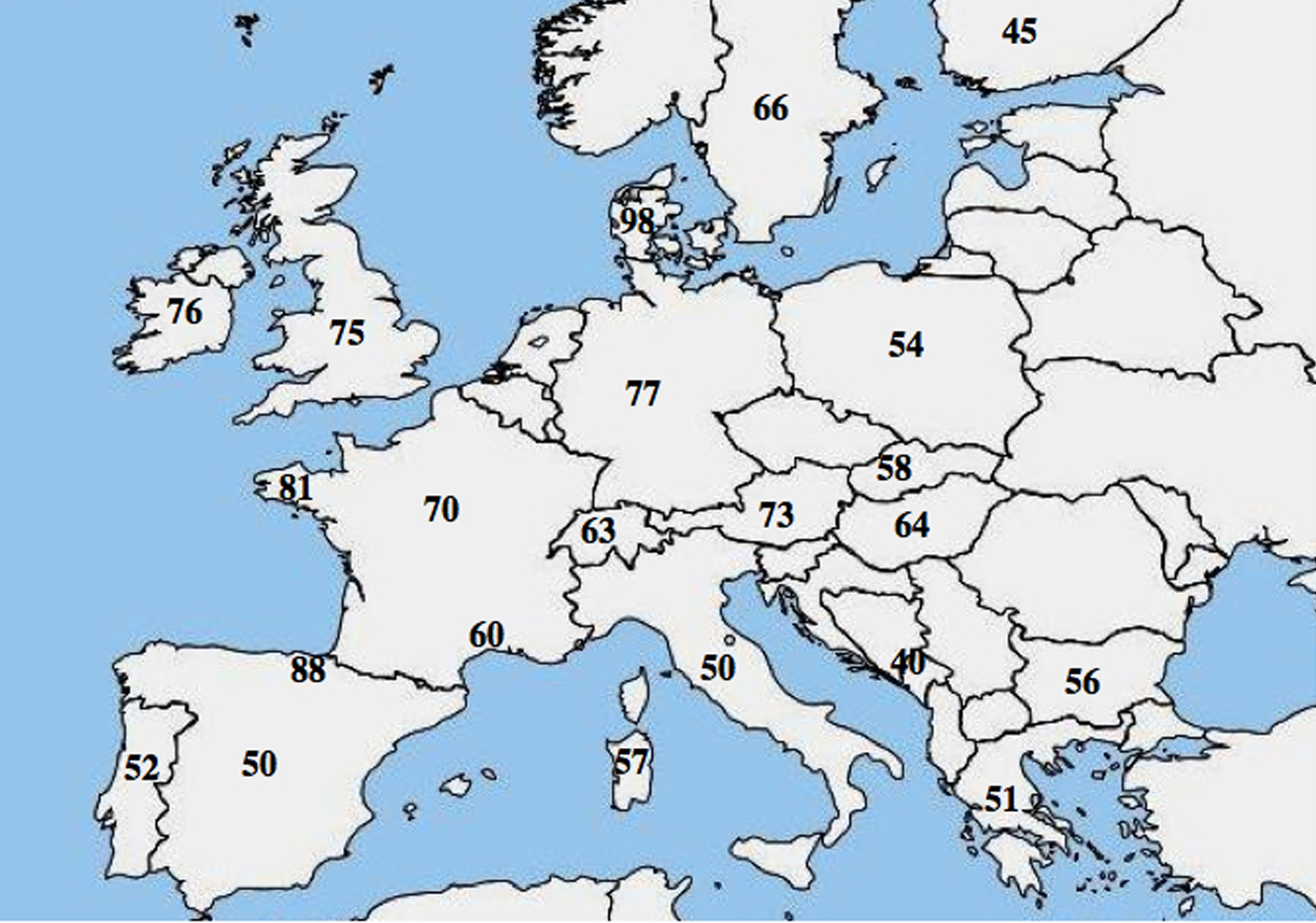
Foi relatada uma heterogeneidade alélica molecular no CF; numerosos estudos destacaram uma diferença acentuada na freqüência do p.Phe508del entre as populações e mostraram um gradiente decrescente de noroeste para sudeste na Europa.
Classes of CFTR mutations
Mutações no gene CFTR podem ser classificadas em seis classes de acordo com seus mecanismos moleculares e conseqüências para diferentes aspectos da biogênese, metabolismo e função do CFTR.
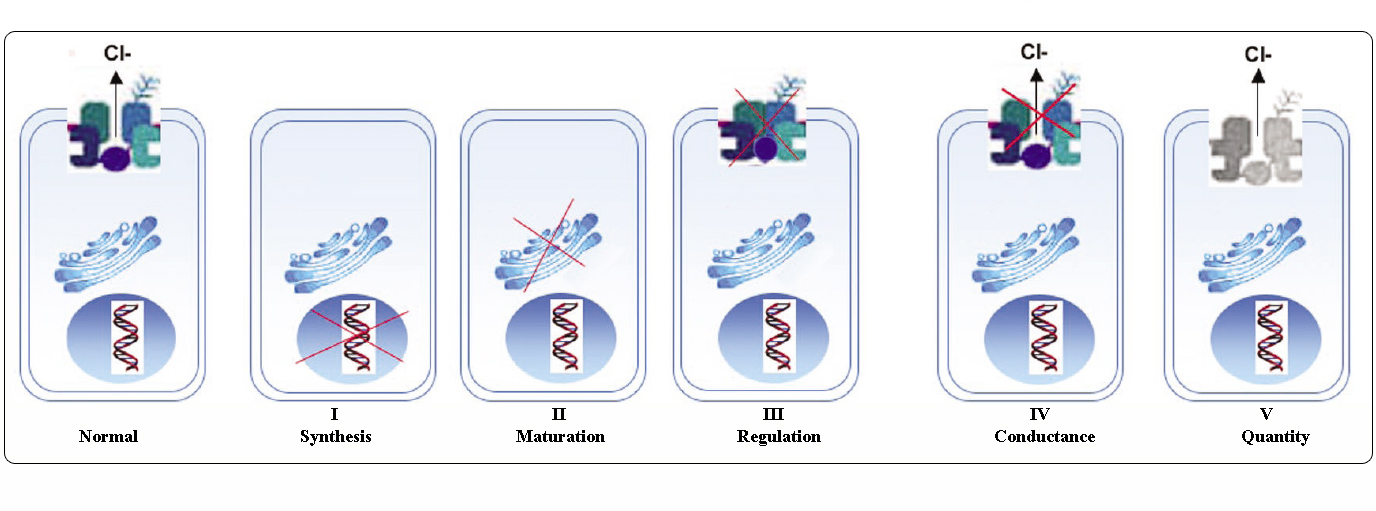
Adaptado de Welsh and Smith (1993) e modificado de Claustres (RBM online, 2005)
Mutações pertencentes às classes I-III e VI conferem pouco ou nenhum CFTR funcional na membrana apical e são consideradas “graves” e geralmente levam a um fenótipo clássico de CF com insuficiência pancreática (CF-PI) embora a gravidade da doença pulmonar possa ser variável.
Mutações pertencentes às classes IV e V retêm alguma atividade residual de CFTR e conferem um fenótipo mais leve. Em pacientes com pelo menos um alelo CF “leve”, a função CFTR é geralmente suficiente para a digestão, e a doença pulmonar é menos grave. Indivíduos com mutantes CFTR que dão origem a cerca de 10% do nível normal de mRNA CFTR podem apresentar apenas um sintoma, como a CBAVD (ausência bilateral congênita de vaso deferente) em indivíduos do sexo masculino. Alguns indivíduos heterozigotos para uma mutação por CFTR podem estar em risco aumentado de “distúrbios relacionados ao CFTR” (CFTR-RD), como pancreatite, sinusite ou aspergilose broncopulmonar alérgica. Nesses casos, as mutações CFTR podem ser um fator de predisposição genética e modificadores genéticos ou ambientais podem ter efeitos aditivos.
O agrupamento das mutações em diferentes classes é útil para entender o mecanismo da disfunção, entretanto muitas variações de seqüência são raras/únicas e os dados disponíveis não permitem avaliar sua patogenicidade. Além disso, uma única mutação pode causar mais de um tipo de anormalidade ou pode conferir um fenótipo variável.
* Claustres M. 2005. Patologia molecular do locus CFTR na infertilidade masculina. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Complexidade em uma doença monogênica. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identificação do gene da fibrose cística: análise genética. Ciência 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL e outros. 1989. Identificação do gene da fibrose cística: clonagem e caracterização do ADN complementar. Ciência 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Localização física de dois marcadores de DNA intimamente ligados ao locus de fibrose cística por eletroforese em gel de campo pulsado. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. Mecanismos moleculares da disfunção do canal de cloreto CFTR na fibrose cística. Célula 73(7):1251-4. PMID: 768686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genoma 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. Variante do gene CFTR para pacientes com ausência congênita de vaso deferente. Am J Hum Genet 57(4):958-60. PMID: 7573058.