 |
Kystisen fibroosin transmembraanisen konduktanssin säätelijägeeni (CFTR, OMIM #602421) kuuluu ATP:tä sitovien kasettien C-alatyypin (ABCC7) alatyypin perheeseen. Geeni, joka löydettiin vuonna 1989 (Kerem et al., 1989; Riordan et al., 1989; Rommens et al., 1989), ulottuu 188 702 bp:n alueelle kromosomi 7:n pitkällä haaralla (kaistat 7q31-q32) (sijainti 117 119 358-117 308 719) ja koostuu 27 eksonista, jotka alun perin numeroitiin 1-24 ja joissa on alajaottelu a ja b eksoneille 6, 14 ja 17 (Zielenski et al., 1991). Kypsän transkriptin pituus on 6 129 nukleotidia, mukaan lukien 4 440 koodaavan emäksen avoin lukukehys (viitesekvenssi: NM_000492). Ensembl Gene ID: ENSG00000001626 Kromosomi 7 ( NCBI, tai HGNC) |
Nimikkeistö
UMD-CFTR käyttää suositeltuja nimikkeistöohjeita, joita Human Genome Variation Society (www.hgvs.org/mutnomen/) ehdottaa esim, cDNA-pohjainen numerointi, jossa ATG-translaation aloituskodonin A on +1 ja eksonit numeroidaan 1-27.
UMD-CFTR:ssä koodaava referenssisekvenssi on NM_000492 lukuun ottamatta c.1408A:ta G:n sijasta ja referenssiproteiinisekvenssi on NP_000483 lukuun ottamatta p.Met470:tä p.Val470:n sijasta.
Kystisen fibroosin mutaatiotietokantakonsortioiden (www.genet.sickkids.on.ca/cftr/) numerointitapaan (cystic fibrosis mutations database consortium consortium numeeraus, cystic fibrosis mutations database consortium numbering, www.genet.sickkids.on.ca/cftr/) tottuneille käyttäjille on tarjottu kahdet imuroitavat tiedostot:
– eksonien ja nukleotidien vastaavuusnumerointi: yleinen numerointi (valkoisella eksonien osalta ja mustalla nukleotidien osalta) ja suositeltu numerointi (punaisella). Kunkin eksonin ensimmäinen ja viimeinen nukleotidi on merkitty. (download) (Show/Hide)
– muunnosten nimien vastaavuustaulukko. (download)
Koodaava sekvenssi (Show/Hide)
CFTR-mutaatiot
Lähes 1700 CFTR-sekvenssin mutaatiota ja variaatiota on tähän mennessä kuvattu (www.genet.sickkids.on.ca/cftr). Maailmanlaajuisesti yleisin kystiseen fibroosiin johtava mutaatio on kolmen emäksen deleetio, joka koodaa fenyylialaniinijäämää positiossa 508 nimellä p.Phe508del, mikä heikentää CFTR-proteiinin kykyä taittua endoplasmisessa retikulumissa (ER), mikä lisää proteiinin nopeaa hajoamista ER-prosessoinnin aikana. Sen osuus CF-alleeleista on noin 70 prosenttia.
Täyspitkän p.Phe508del-CFTR-cDNA:n heterologinen ilmentäminen on osoittanut, että tämä mutaatioproteiini saa vain ydinglykosylaation eikä monimutkaisia oligosakkaridiketjuja eikä näin ollen pääse kulkeutumaan solun pinnalle, mikä vastaa havaintoa, jonka mukaan mutanttiproteiinia ei esiinny potilailta saaduissa tuoreissa kudosten epiteeleissä. Muiden haitallisten vaikutusten ohella p.Phe508del heikentää CFTR:n oikeaa taittumista ja peittää myös konformaatiomielessä NBD1:n kolmen jäännöksen diasidisen poistumiskoodin, joka näyttää olevan välttämätön ER:n viennille.
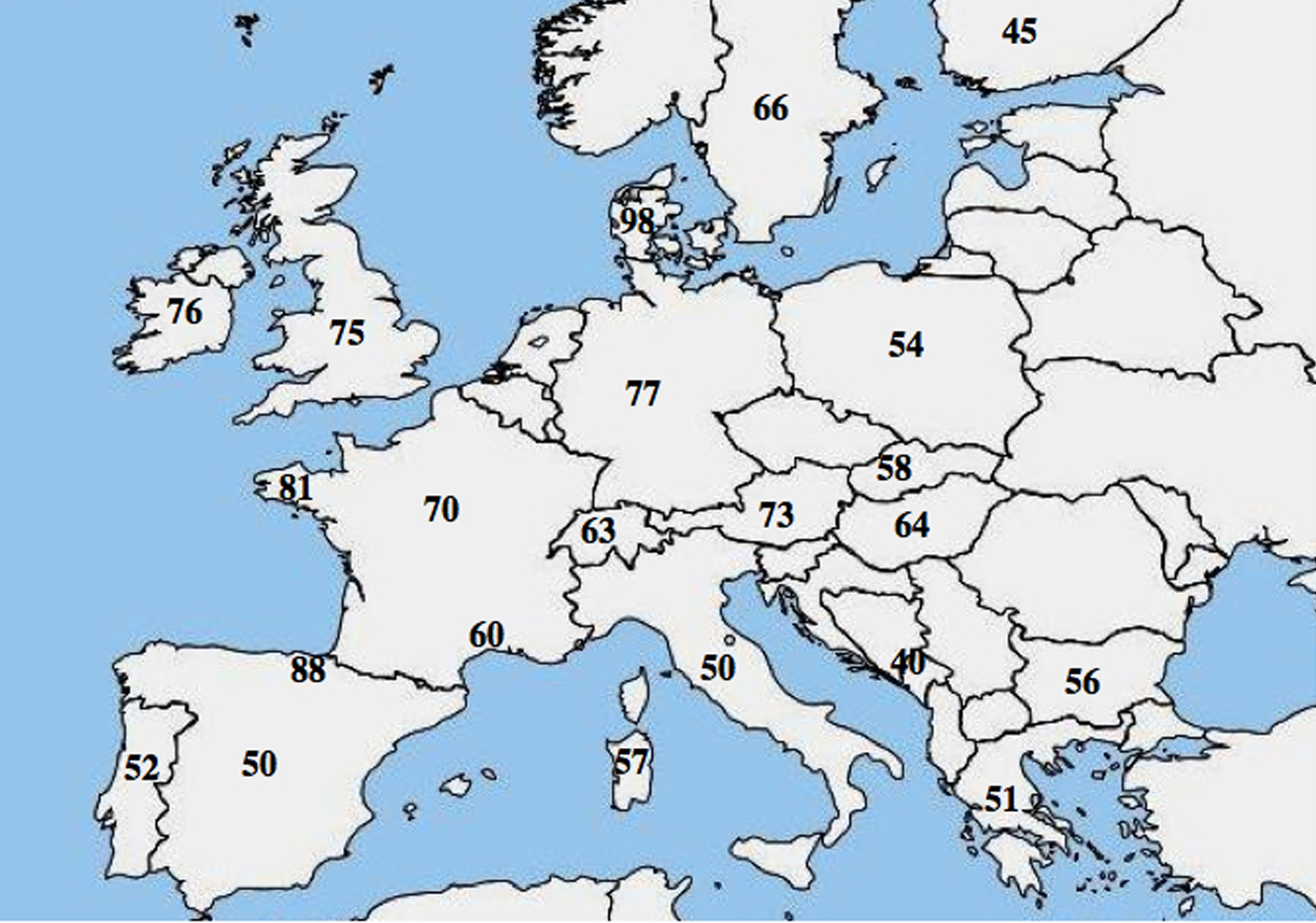
Molekulaarisesta alleelisesta heterogeenisyydestä CF:ssä on raportoitu; lukuisat tutkimukset ovat osoittaneet huomattavia eroja populaatioiden välisessä esiintyvyydessä, ja ne ovat osoittaneet, että luoteesta kaakkoon suuntautuva gradientti on pienentynyt Euroopassa.
CFTR-mutaatioiden luokat
CFTR-geenin mutaatiot voidaan luokitella kuuteen luokkaan niiden molekulaaristen mekanismien ja CFTR:n biogeneesin, metabolian ja toiminnan eri näkökohtiin kohdistuvien seurausten mukaan.
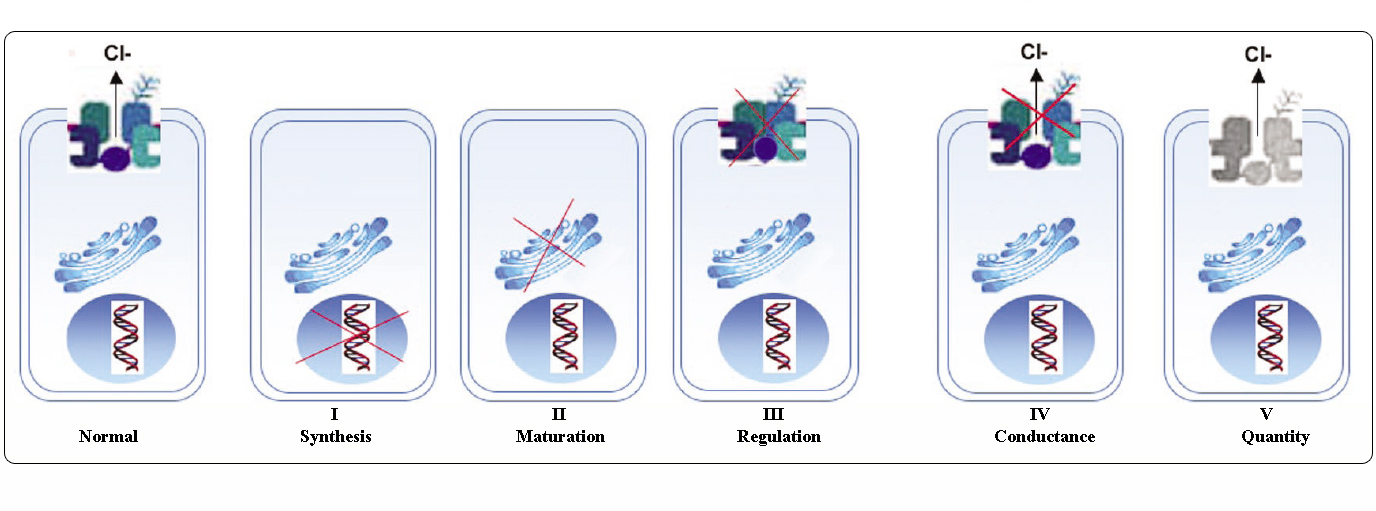
Sovitettu lähteestä Welsh ja Smith (1993) ja muokattu lähteestä Claustres (RBM online, 2005)
Luokkiin I-III ja VI kuuluvat mutaatiot aiheuttavat vain vähän tai ei lainkaan funktionaalista CFTR:ää apikaalikalvolla, ja niitä pidetään ”vakavina”, ja ne johtavat tavallisesti klassiseen CF-fenotyyppiin, johon liittyy haiman vajaatoiminta (CF-PI), vaikkakin keuhko- ja hengityselinten sairauden vaikeusaste voi vaihdella.
Luokkiin IV ja V kuuluvissa mutaatioissa säilyy jonkin verran jäljellä olevaa CFTR-aktiivisuutta, ja ne aiheuttavat lievemmän fenotyypin. Potilailla, joilla on vähintään yksi ”lievä” CF-alleeli, CFTR-toiminta on yleensä riittävä ruoansulatuksen kannalta, ja keuhkosairaus on lievempi. Henkilöillä, joilla on CFTR-mutaatioita, jotka synnyttävät noin 10 % normaalista CFTR-mRNA:sta, voi esiintyä vain yksi oire, kuten CBAVD (synnynnäinen molemminpuolinen vas deferensin puuttuminen) miehillä. Joillakin henkilöillä, joilla on heterotsygootti CFTR-mutaatio, saattaa olla kohonnut riski sairastua ”CFTR:ään liittyviin häiriöihin” (CFTR-RD), kuten haimatulehdukseen, poskiontelotulehdukseen tai allergiseen bronkopulmonaaliseen aspergilloosiin. Näissä tapauksissa CFTR-mutaatiot voivat olla geneettisen alttiuden tekijä, ja geneettisillä tai ympäristöön liittyvillä tekijöillä voi olla additiivisia vaikutuksia.
Mutaatioiden ryhmittely eri luokkiin on hyödyllistä toimintahäiriön mekanismin ymmärtämiseksi, mutta monet sekvenssivariaatiot ovat harvinaisia/yksilöllisiä, eikä niiden patogeenisuutta voida käytettävissä olevien tietojen perusteella arvioida. Lisäksi yksittäinen mutaatio voi aiheuttaa useamman kuin yhden tyyppisiä poikkeavuuksia tai aiheuttaa vaihtelevan fenotyypin.
* Claustres M. 2005. CFTR-lookuksen molekyylipatologia miesten hedelmättömyydessä. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Monogeenisen sairauden monimutkaisuus. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Kystisen fibroosin geenin tunnistaminen: geneettinen analyysi. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL ja muut. 1989. Kystisen fibroosin geenin tunnistaminen: komplementaarisen DNA:n kloonaus ja karakterisointi. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Kahden kystisen fibroosin lokukseen läheisesti liittyvän DNA-markkerin fyysinen lokalisaatio pulssikenttägeelielektroforeesilla. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. CFTR-kloridikanavan toimintahäiriön molekyylimekanismit kystisessä fibroosissa. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Kystisen fibroosin transmembraanisen konduktanssin säätäjän (CFTR) geenin genominen DNA-sekvenssi. Genomics 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. CFTR-geenivariantti potilailla, joilla on synnynnäinen vas deferensin puuttuminen. Am J Hum Genet 57(4):958-60. PMID: 7573058.