 |
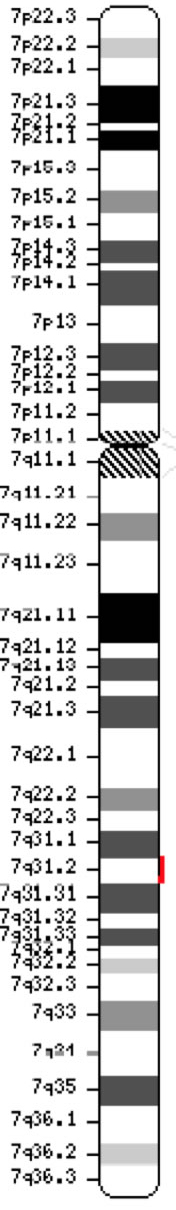
Het cystische fibrose transmembraan geleidingsregulator-gen (CFTR, OMIM #602421) is een lid van de ATP-bindende cassette sub-familie C (ABCC7). Het gen, dat in 1989 werd ontdekt (Kerem et al, 1989; Riordan et al, 1989; Rommens et al, 1989), beslaat 188.702 bp op de lange arm van chromosoom 7 (band 7q31-q32) (positie 117.119.358-117.308.719) en is samengesteld uit 27 exonen die aanvankelijk genummerd waren als 1-24 met onderverdelingen a en b voor exonen 6, 14 en 17 (Zielenski et al., 1991). Het rijpe transcript is 6.129 nucleotiden lang, inclusief een open leesraam van 4.440 coderende basen (Referentiesequentie: NM_000492). Ensembl Gene ID: ENSG00000001626 Chromosoom 7 ( NCBI, of HGNC) |
Nomenclatuur
Het UMD-CFTR gebruikt de aanbevolen nomenclatuurrichtlijnen die zijn voorgesteld door de Human Genome Variation Society (www.hgvs.org/mutnomen/), bijv, cDNA-gebaseerde nummering met de A van ATG translationeel initiatiecodon op +1 en exonen genummerd van 1 tot 27.
In de UMD-CFTR is de coderende referentiesequentie NM_000492 met uitzondering van c.1408A in plaats van G, en de referentie-eiwitsequentie is NP_000483 met uitzondering van p.Met470 in plaats van p.Val470.
Voor het gemak van de gebruikers die gewend zijn aan de cystische fibrose mutaties database consortium nummering (www.genet.sickkids.on.ca/cftr/) twee downloadbare bestanden worden verstrekt:
– een corresponderende nummering voor exonen en nucleotiden: gangbare nummering (in wit voor exonen en in zwart voor nucleotiden) en aanbevolen nummering (in rood). Voor elk exon worden de eerste en de laatste nucleotide aangegeven. (download) (Weergeven/Verbergen)
– een correspondentietabel voor variatienamen. (download)
Coding sequence (Show/Hide)
De CFTR-mutaties
Al bijna 1.700 CFTR-sequentiemutaties en -variaties zijn tot nu toe beschreven (www.genet.sickkids.on.ca/cftr). De wereldwijd meest voorkomende mutatie die verantwoordelijk is voor cystische fibrose is een deletie van drie basen die codeert voor een fenylalanineresidu op positie 508, p.Phe508del genaamd, die het vermogen van het CFTR-eiwit om zich in het endoplasmatisch reticulum (ER) te vouwen belemmert, waardoor de snelle degradatie van het eiwit tijdens ER-verwerking wordt bevorderd. Het maakt ongeveer 70% van de CF-allelen uit.
Heterologe expressie van full-length p.Phe508del-CFTR-cDNA heeft aangetoond dat dit mutante eiwit alleen kernglycosylering verkrijgt en geen complexe oligosaccharideketens en als zodanig niet naar het celoppervlak wordt getransporteerd, in overeenstemming met de waarneming van afwezigheid van mutant-eiwit in verse epithelia van patiëntenweefsels. Naast andere schadelijke effecten belemmert p.Phe508del de correcte vouwing van CFTR en maskeert het ook conformationeel de diacidische exit code van drie residuen in de NBD1 die noodzakelijk lijkt voor ER export.
Er is een moleculaire allel heterogeniteit in CF gerapporteerd; talrijke studies wezen op een duidelijk verschil in de frequentie van p.Phe508del tussen populaties en toonden een afnemende noordwestelijke naar zuidoostelijke gradiënt in Europa.
Klassen CFTR-mutaties
Mutaties in het CFTR-gen kunnen in zes klassen worden ingedeeld op basis van hun moleculaire mechanismen en gevolgen voor verschillende aspecten van de biogenese, het metabolisme en de functie van CFTR.
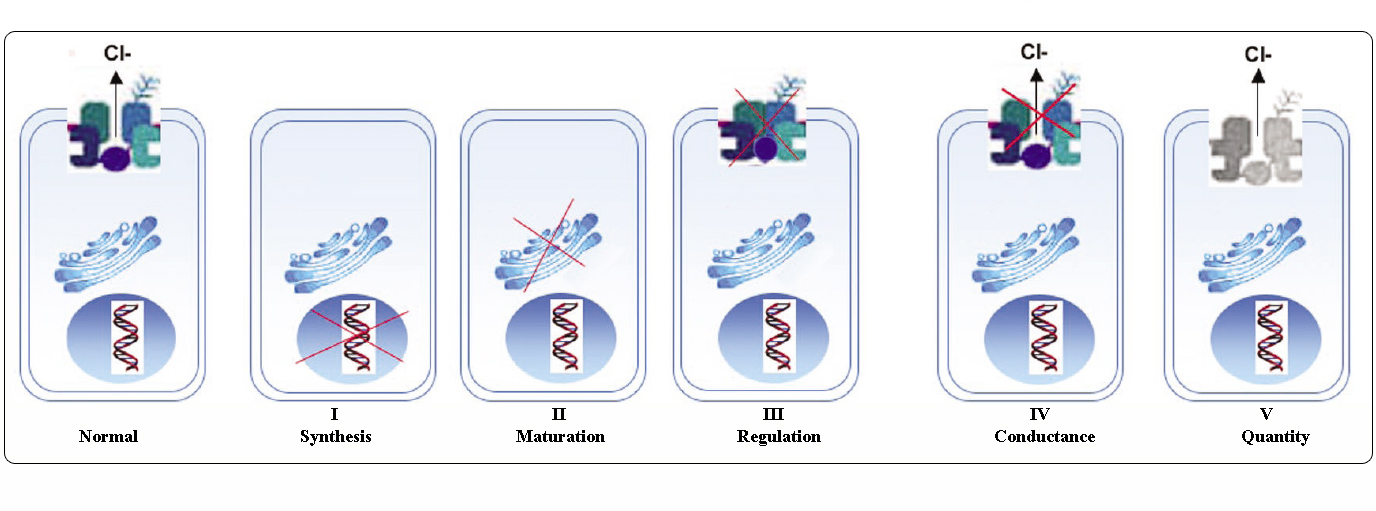
Opgesteld op basis van Welsh en Smith (1993) en gewijzigd op basis van Claustres (RBM online, 2005)
Mutaties die tot de klassen I-III en VI behoren, geven weinig of geen functioneel CFTR aan het apicale membraan en worden als “ernstig” beschouwd en leiden meestal tot een klassiek CF-fenotype met pancreasinsufficiëntie (CF-PI), hoewel de ernst van de longaandoening variabel kan zijn.
Mutaties die tot de klassen IV en V behoren, behouden enige residuele CFTR-activiteit en geven een milder fenotype. Bij patiënten met ten minste één “mild” CF-allel is de CFTR-functie meestal voldoende voor de spijsvertering, en is de longaandoening minder ernstig. Personen met CFTR-mutanten die ongeveer 10% van het normale niveau van CFTR-mRNA veroorzaken, kunnen zich met slechts één symptoom presenteren, zoals CBAVD (congenitale bilaterale afwezigheid van zaadleiders) bij mannen. Sommige personen die heterozygoot zijn voor een CFTR-mutatie, lopen een verhoogd risico op “CFTR-gerelateerde aandoeningen” (CFTR-RD), zoals pancreatitis, sinusitis of allergische bronchopulmonale aspergillose. In deze gevallen zouden CFTR-mutaties een factor van genetische aanleg kunnen zijn en zouden genetische of omgevingsmodifiers een additief effect kunnen hebben.
Het groeperen van mutaties in verschillende klassen is nuttig om het mechanisme van disfunctie te begrijpen, maar veel sequentievariaties zijn zeldzaam/uniek en de beschikbare gegevens laten niet toe om hun pathogeniciteit te beoordelen. Bovendien kan een enkele mutatie meer dan een type afwijking veroorzaken of een variabel fenotype geven.
* Claustres M. 2005. Moleculaire pathologie van de CFTR locus in mannelijke onvruchtbaarheid. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Complexiteit in een monogene ziekte. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identificatie van het cystic fibrosis gen: genetische analyse. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL en anderen. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Physical localization of two DNA markers closely linked to the cystic fibrosis locus by pulsed-field gel electrophoresis. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. Moleculaire mechanismen van CFTR chloride kanaal disfunctie in cystic fibrosis. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. CFTR genvariant voor patiënten met congenitale afwezigheid van vas deferens. Am J Hum Genet 57(4):958-60. PMID: 7573058.