 |
Le gène du régulateur de conductance transmembranaire de la fibrose kystique (CFTR, OMIM #602421) est un membre de la sous-famille C des cassettes de liaison à l’ATP (ABCC7). Le gène, découvert en 1989 (Kerem et al, 1989 ; Riordan et al, 1989 ; Rommens et al, 1989), s’étend sur 188 702 pb sur le bras long du chromosome 7 (bande 7q31-q32) (position 117 119 358-117 308 719) et est composé de 27 exons initialement numérotés de 1 à 24 avec des subdivisions a et b pour les exons 6, 14 et 17 (Zielenski et al, 1991). Le transcrit mature est long de 6 129 nucléotides incluant un cadre de lecture ouvert de 4 440 bases codantes (Séquence de référence : NM_000492). Ensembl Gene ID : ENSG00000001626 Chromosome 7 ( NCBI, ou HGNC) |
Nomenclature
L’UMD-CFTR utilise les directives de nomenclature recommandées proposées par la Human Genome Variation Society (www.hgvs.org/mutnomen/), par exemple, numérotation basée sur l’ADNc avec le A du codon d’initiation de la traduction ATG à +1 et les exons numérotés de 1 à 27.
Dans l’UMD-CFTR, la séquence de référence codante est NM_000492 à l’exception de c.1408A au lieu de G, et la séquence protéique de référence est NP_000483 à l’exception de p.Met470 au lieu de p.Val470.
Pour faciliter la tâche des utilisateurs habitués à la numérotation du consortium de la base de données des mutations de la fibrose kystique (www.genet.sickkids.on.ca/cftr/), deux fichiers téléchargeables sont fournis :
– une numérotation de correspondance pour les exons et les nucléotides : numérotation commune (en blanc pour les exons et en noir pour les nucléotides) et numérotation recommandée (en rouge). Le premier et le dernier nucléotide sont indiqués pour chaque exon. (download) (Show/Hide)
– une table de correspondance pour les noms des variations. (download)
Séquence codante (Show/Hide)
Les mutations CFTR
Près de 1 700 mutations et variations de la séquence CFTR ont été décrites à ce jour (www.genet.sickkids.on.ca/cftr). La mutation la plus fréquente dans le monde et responsable de la mucoviscidose est une délétion de trois bases codant pour un résidu phénylalanine en position 508 nommée p.Phe508del, qui altère la capacité de la protéine CFTR à se replier dans le réticulum endoplasmique (RE), favorisant ainsi la dégradation rapide de la protéine lors du traitement du RE. Il représente environ 70 % des allèles de CF.
L’expression hétérologue de l’ADNc p.Phe508del-CFTR complet a démontré que cette protéine mutante n’acquiert qu’une glycosylation de base et non des chaînes oligosaccharidiques complexes et, de ce fait, ne parvient pas à être transportée à la surface cellulaire, conformément à l’observation de l’absence de la protéine mutante dans les épithéliums frais provenant de tissus de patients. Parmi d’autres effets néfastes, p.Phe508del entrave le repliement correct de CFTR et masque également de manière conformationnelle le code de sortie diacide de trois résidus dans le NBD1 qui semble nécessaire pour l’exportation vers le RE.
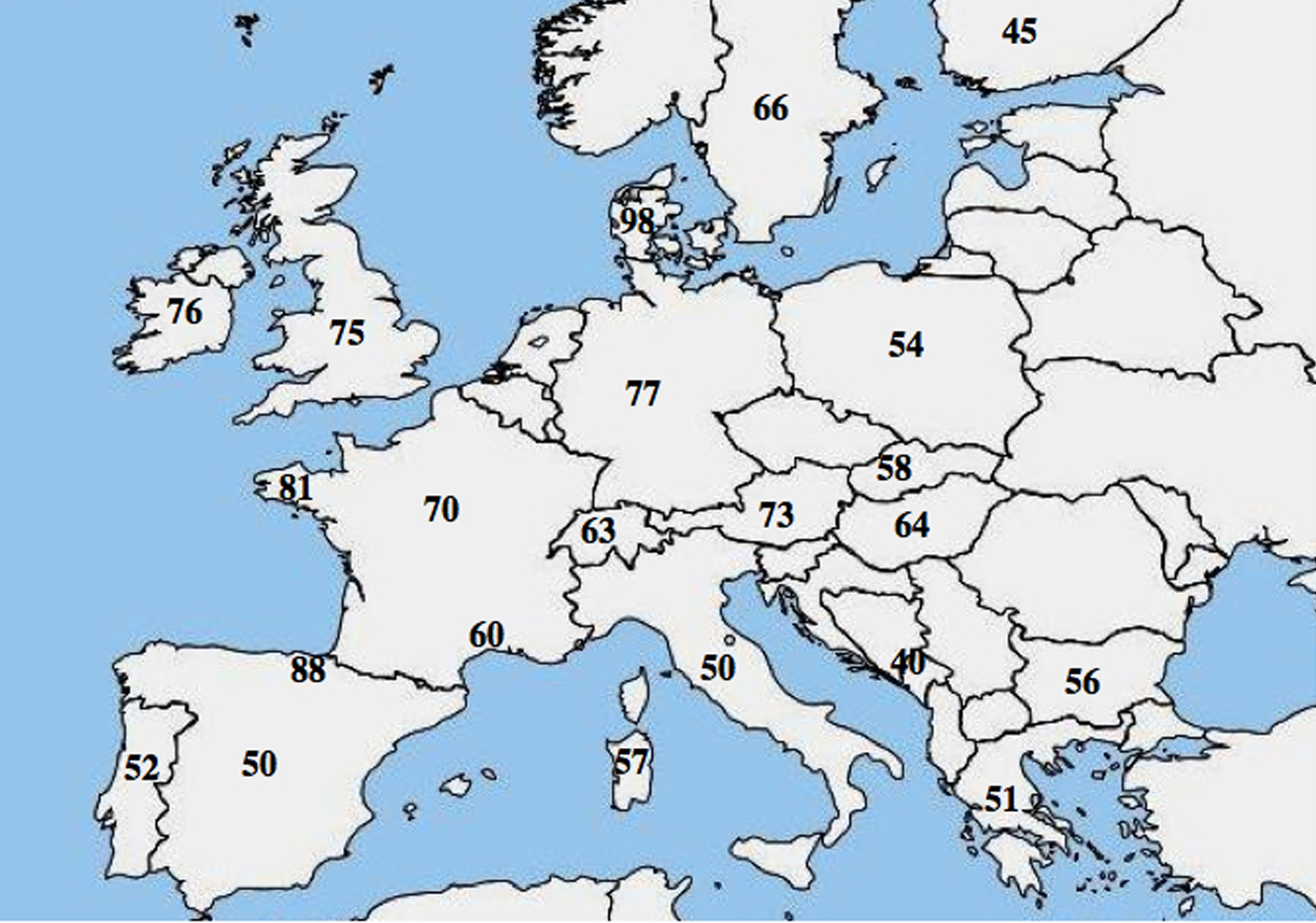
Une hétérogénéité allélique moléculaire dans la CF a été rapportée ; de nombreuses études ont mis en évidence une différence marquée dans la fréquence de p.Phe508del parmi les populations et ont montré un gradient décroissant du nord-ouest au sud-est en Europe.
Classes de mutations CFTR
Les mutations du gène CFTR peuvent être classées en six classes selon leurs mécanismes moléculaires et leurs conséquences sur différents aspects de la biogenèse, du métabolisme et de la fonction de CFTR.
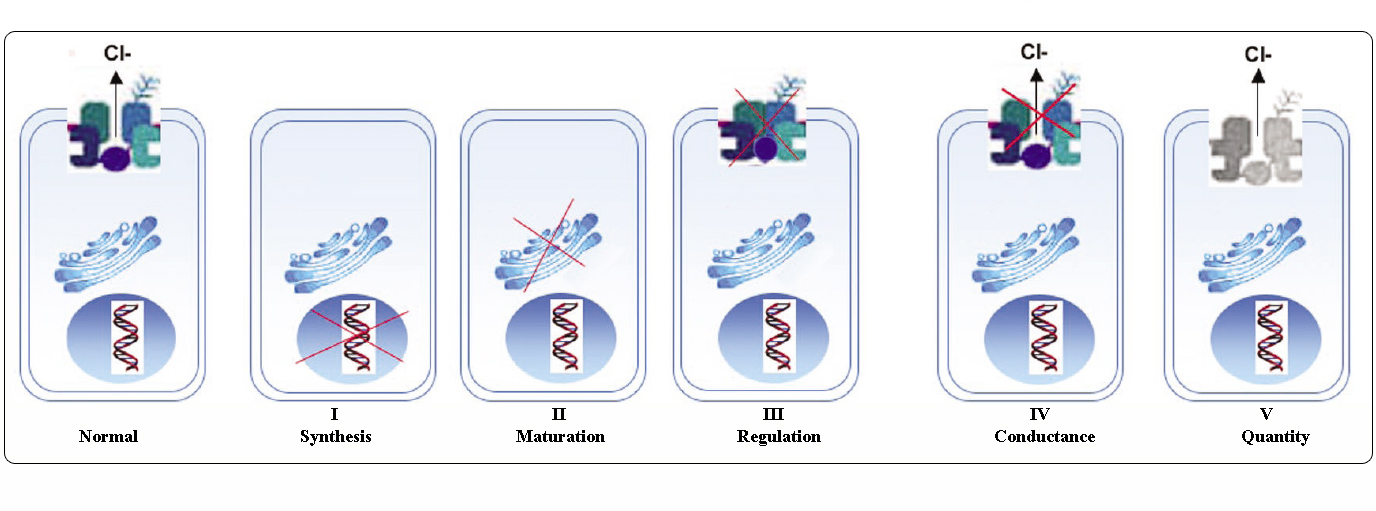
Adapté de Welsh et Smith (1993) et modifié de Claustres (RBM online, 2005)
Les mutations appartenant aux classes I-III et VI confèrent peu ou pas de CFTR fonctionnel à la membrane apicale et sont considérées comme « sévères » et conduisent généralement à un phénotype CF classique avec insuffisance pancréatique (CF-PI) bien que la sévérité de la maladie pulmonaire puisse être variable.
Les mutations appartenant aux classes IV et V conservent une certaine activité CFTR résiduelle et confèrent un phénotype plus léger. Chez les patients présentant au moins un allèle CF « léger », la fonction CFTR est généralement suffisante pour la digestion, et la maladie pulmonaire est moins grave. Les individus porteurs de mutants CFTR qui donnent lieu à environ 10 % du niveau normal d’ARNm CFTR peuvent ne présenter qu’un seul symptôme, comme l’absence bilatérale congénitale des canaux déférents (CBAVD) chez les hommes. Certains individus hétérozygotes pour une mutation du CFTR peuvent présenter un risque accru de « troubles liés au CFTR » (CFTR-RD), comme la pancréatite, la sinusite ou l’aspergillose broncho-pulmonaire allergique. Dans ces cas, les mutations de CFTR pourraient être un facteur de prédisposition génétique et les modificateurs génétiques ou environnementaux pourraient jouer des effets additifs.
Grouper les mutations en différentes classes est utile pour comprendre le mécanisme de dysfonctionnement, cependant de nombreuses variations de séquence sont rares/uniques et les données disponibles ne permettent pas d’évaluer leur pathogénicité. De plus, une seule mutation peut causer plus d’un type d’anomalie ou peut conférer un phénotype variable.
* Claustres M. 2005. Pathologie moléculaire du locus CFTR dans l’infertilité masculine. Reprod Biomed Online 10(1):14-41. PMID : 15705292.
* Estivill X. 1996. Complexité dans une maladie monogénique. Nat Genet 12(4):348-50. PMID : 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identification du gène de la fibrose kystique : analyse génétique. Science 245(4922):1073-80. PMID : 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL et autres. 1989. Identification du gène de la fibrose kystique : clonage et caractérisation de l’ADN complémentaire. Science 245(4922):1066-73. PMID : 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Localisation physique de deux marqueurs d’ADN étroitement liés au locus de la fibrose kystique par électrophorèse sur gel à champ pulsé. Am J Hum Genet 45(6):932-41. PMID : 2589321.
* Welsh MJ, Smith AE. 1993. Mécanismes moléculaires du dysfonctionnement du canal chlorure CFTR dans la mucoviscidose. Cell 73(7):1251-4. PMID : 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Séquence d’ADN génomique du gène du régulateur de conductance transmembranaire de la fibrose kystique (CFTR). Genomics 10(1):214-28. PMID : 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. Variante du gène CFTR chez les patients présentant une absence congénitale de canal déférent. Am J Hum Genet 57(4):958-60. PMID : 7573058.