 |
Das CFTR-Gen (Cystic Fibrosis Transmembrane Conductance Regulator, OMIM #602421) gehört zur ATP-bindenden Kassetten-Unterfamilie C (ABCC7). Das 1989 entdeckte Gen (Kerem et al., 1989; Riordan et al., 1989; Rommens et al., 1989) erstreckt sich über 188.702 bp auf dem langen Arm von Chromosom 7 (Band 7q31-q32) (Position 117.119.358-117.308.719) und besteht aus 27 Exons, die ursprünglich mit 1-24 nummeriert waren, mit Unterteilungen a und b für die Exons 6, 14 und 17 (Zielenski et al., 1991). Das reife Transkript ist 6.129 Nukleotide lang, einschließlich eines offenen Leserasters von 4.440 kodierenden Basen (Referenzsequenz: NM_000492). Ensembl Gene ID: ENSG00000001626 Chromosom 7 ( NCBI, oder HGNC) |
Nomenklatur
Das UMD-CFTR verwendet die von der Human Genome Variation Society (www.hgvs.org/mutnomen/) empfohlenen Nomenklatur-Richtlinien, z.B., cDNA-basierte Nummerierung mit dem A des ATG-Translationsinitiationscodons bei +1 und den Exons mit den Nummern 1 bis 27.
In der UMD-CFTR ist die kodierende Referenzsequenz NM_000492 mit Ausnahme von c.1408A anstelle von G, und die Referenzproteinsequenz ist NP_000483 mit Ausnahme von p.Met470 anstelle von p.Val470.
Zur Erleichterung der Benutzer, die an die Nummerierung der Cystic Fibrosis Mutations Database Consortium (www.genet.sickkids.on.ca/cftr/) gewöhnt sind, werden zwei herunterladbare Dateien bereitgestellt:
– eine korrespondierende Nummerierung für Exons und Nukleotide: übliche Nummerierung (in weiß für Exons und in schwarz für Nukleotide) und empfohlene Nummerierung (in rot). Die ersten und letzten Nukleotide sind für jedes Exon angegeben. (download) (Anzeigen/Verbergen)
– eine Korrespondenztabelle für Variationsnamen. (download)
Kodierende Sequenz (Einblenden/Verbergen)
Die CFTR-Mutationen
Fast 1.700 CFTR-Sequenzmutationen und -Variationen sind bisher beschrieben worden (www.genet.sickkids.on.ca/cftr). Die weltweit häufigste Mutation, die für Mukoviszidose verantwortlich ist, ist eine Deletion von drei Basen, die für einen Phenylalaninrest an Position 508 kodiert (p.Phe508del), der die Fähigkeit des CFTR-Proteins beeinträchtigt, sich im endoplasmatischen Retikulum (ER) zu falten, wodurch der schnelle Abbau des Proteins während der ER-Verarbeitung gefördert wird. Sie macht etwa 70 % der CF-Allele aus.
Die heterologe Expression von p.Phe508del-CFTR-cDNA in voller Länge hat gezeigt, dass dieses mutierte Protein nur eine Kernglykosylierung und keine komplexen Oligosaccharidketten erhält und daher nicht an die Zelloberfläche transportiert werden kann, was mit der Beobachtung übereinstimmt, dass das mutierte Protein in frischen Epithelien von Patientengeweben fehlt. Neben anderen nachteiligen Effekten beeinträchtigt p.Phe508del die korrekte Faltung von CFTR und maskiert auch konformativ den diacidischen Exit-Code von drei Resten in der NBD1, der für den ER-Export notwendig zu sein scheint.
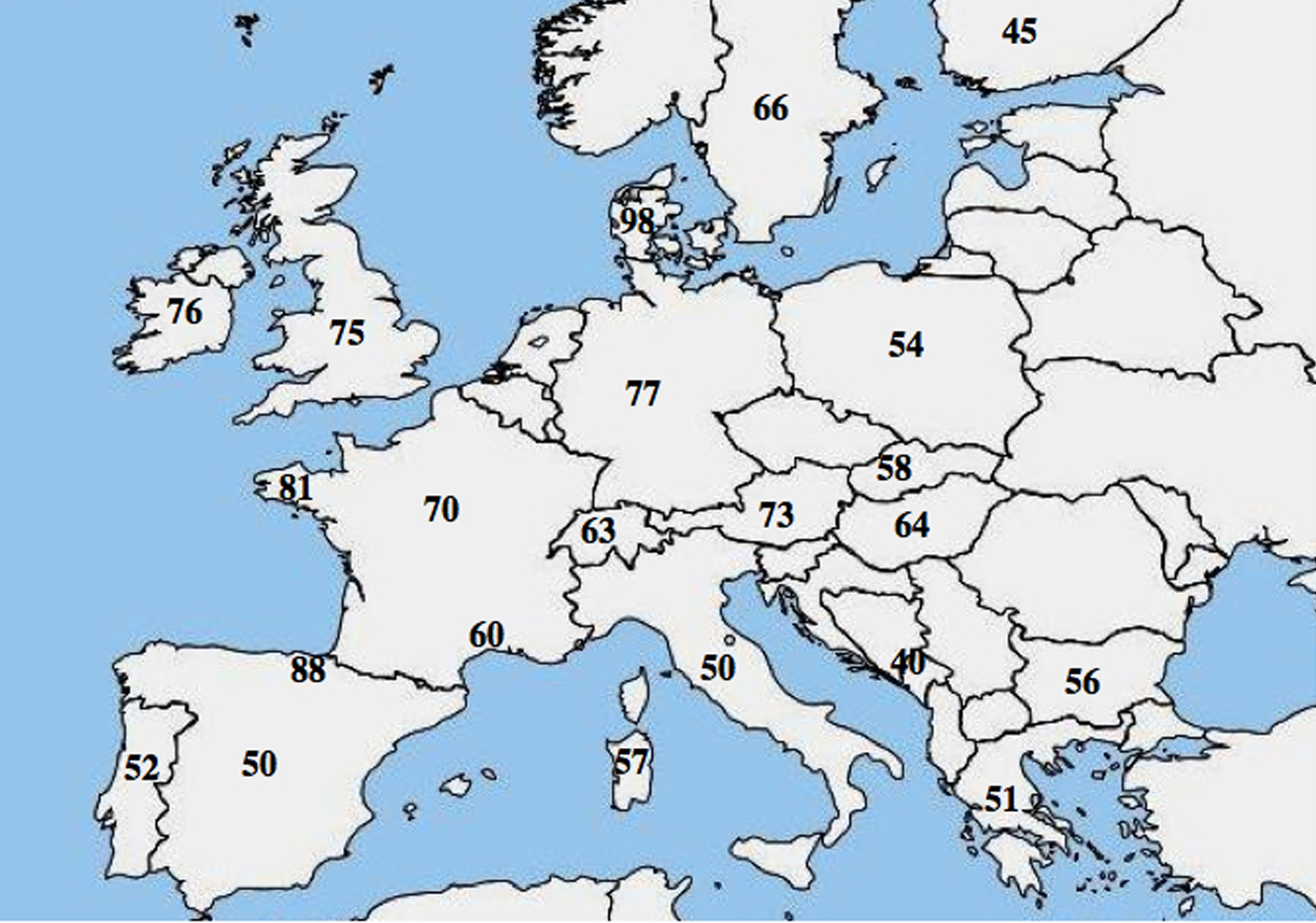
Eine molekulare allelische Heterogenität bei Mukoviszidose wurde berichtet; zahlreiche Studien haben einen deutlichen Unterschied in der Häufigkeit von p.Phe508del zwischen den Populationen hervorgehoben und ein abnehmendes Nordwest-Südost-Gefälle in Europa gezeigt.
Klassen von CFTR-Mutationen
Mutationen im CFTR-Gen können nach ihren molekularen Mechanismen und Folgen für verschiedene Aspekte der CFTR-Biogenese, des Stoffwechsels und der Funktion in sechs Klassen eingeteilt werden.
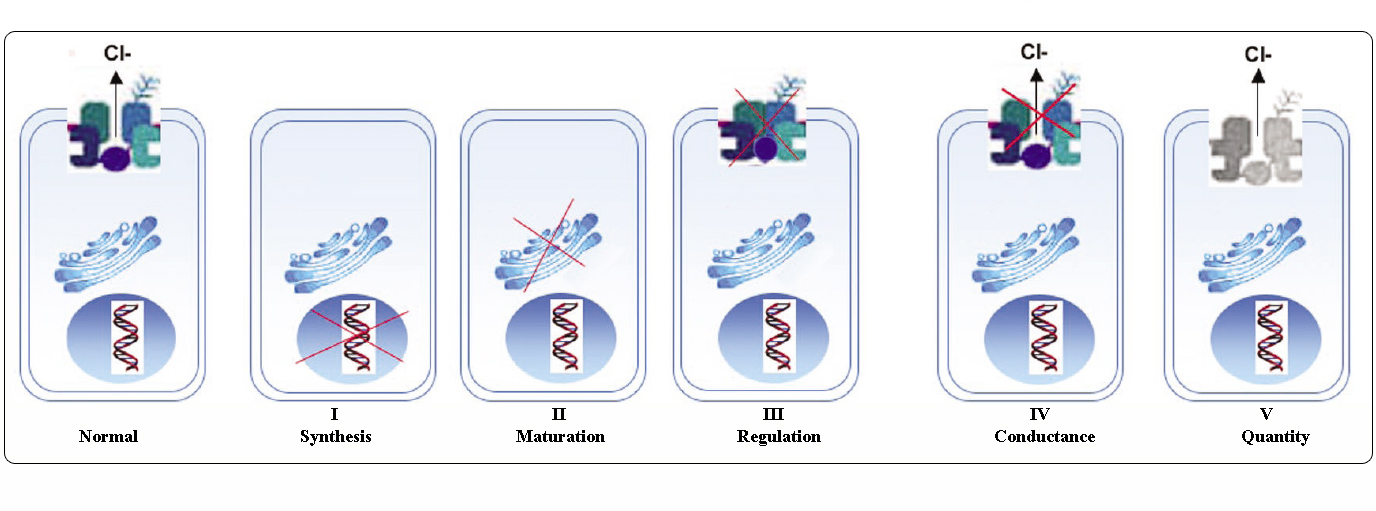
Angepasst von Welsh und Smith (1993) und modifiziert von Claustres (RBM online, 2005)
Mutationen, die zu den Klassen I-III und VI gehören, verleihen wenig oder kein funktionelles CFTR an der apikalen Membran und gelten als „schwer“ und führen in der Regel zu einem klassischen CF-Phänotyp mit Pankreasinsuffizienz (CF-PI), obwohl der Schweregrad der Lungenerkrankung variabel sein kann.
Mutationen, die zu den Klassen IV und V gehören, behalten eine gewisse Restaktivität von CFTR und führen zu einem milderen Phänotyp. Bei Patienten mit mindestens einem „milden“ CF-Allel ist die CFTR-Funktion in der Regel ausreichend für die Verdauung, und die Lungenerkrankung ist weniger schwerwiegend. Personen mit CFTR-Mutanten, die etwa 10 % der normalen Menge an CFTR-mRNA produzieren, können nur ein Symptom aufweisen, wie z. B. CBAVD (congenital bilateral absence of vas deferens) bei Männern. Einige Personen, die heterozygot für eine CFTR-Mutation sind, können ein erhöhtes Risiko für „CFTR-bezogene Störungen“ (CFTR-RD) wie Pankreatitis, Sinusitis oder allergische bronchopulmonale Aspergillose aufweisen. In diesen Fällen könnten CFTR-Mutationen ein Faktor der genetischen Prädisposition sein, und genetische oder umweltbedingte Modifikatoren könnten additive Wirkungen haben.
Die Einteilung von Mutationen in verschiedene Klassen ist nützlich, um den Mechanismus der Funktionsstörung zu verstehen, aber viele Sequenzvariationen sind selten/einzigartig und die verfügbaren Daten erlauben keine Bewertung ihrer Pathogenität. Außerdem kann eine einzige Mutation mehr als eine Art von Anomalie verursachen oder einen variablen Phänotyp verleihen.
* Claustres M. 2005. Molekulare Pathologie des CFTR-Locus bei männlicher Unfruchtbarkeit. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Complexity in a monogenic disease. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identifizierung des Mukoviszidose-Gens: genetische Analyse. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL and others. 1989. Identifizierung des Mukoviszidose-Gens: Klonierung und Charakterisierung der komplementären DNA. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Physikalische Lokalisierung von zwei DNA-Markern, die eng mit dem Mukoviszidose-Locus verbunden sind, durch Impulsfeld-Gelelektrophorese. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. Molekulare Mechanismen der CFTR-Chloridkanal-Dysfunktion bei Mukoviszidose. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Genomische DNA-Sequenz des CFTR-Gens (cystic fibrosis transmembrane conductance regulator). Genomics 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. CFTR-Genvariante bei Patienten mit kongenitalem Fehlen des Vas deferens. Am J Hum Genet 57(4):958-60. PMID: 7573058.