 |
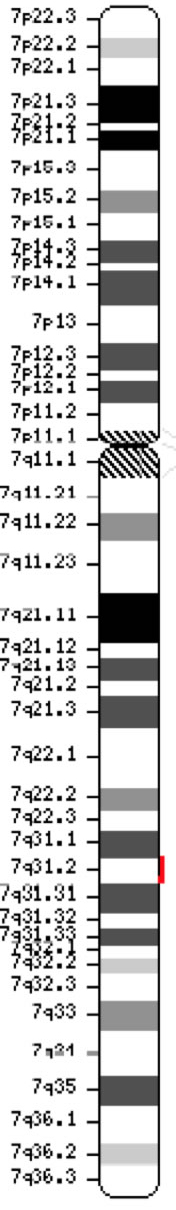
Genetet for cystisk fibrose transmembranledningsregulering (CFTR, OMIM #602421) er et medlem af ATP-binding kassette underfamilie C (ABCC7). Genet, der blev opdaget i 1989 (Kerem et al., 1989; Riordan et al., 1989; Rommens et al., 1989), strækker sig over 188 702 bp på den lange arm af kromosom 7 (bånd 7q31-q32) (position 117 119 358-117 308 719) og består af 27 exoner, der oprindeligt var nummereret 1-24 med underopdeling a og b for exoner 6, 14 og 17 (Zielenski et al., 1991). Det modne transkript er 6.129 nukleotider langt, herunder en åben læseramme på 4.440 kodningsbaser (referencesekvens: NM_000492). Ensembl Gene ID: ENSG00000001626 Chromosom 7 ( NCBI, eller HGNC) |
Nomenklatur
Den UMD-CFTR anvender de anbefalede nomenklaturretningslinjer, der er foreslået af Human Genome Variation Society (www.hgvs.org/mutnomen/), f.eks, cDNA-baseret nummerering med A af ATG-translationsinitieringskodonet ved +1 og exonerne nummereret fra 1 til 27.
I UMD-CFTR er den kodende referencesekvens NM_000492 med undtagelse af c.1408A i stedet for G, og referenceproteinsekvensen er NP_000483 med undtagelse af p.Met470 i stedet for p.Val470.
For at gøre det lettere for de brugere, der er vant til nummereringen i cystic fibrosis mutations database consortium (www.genet.sickkids.on.ca/cftr/), er der to filer, der kan downloades:
– en korrespondancenummerering for exoner og nukleotider: almindelig nummerering (i hvidt for exoner og i sort for nukleotider) og anbefalet nummerering (i rødt). De første og sidste nukleotider er angivet for hvert exon. (download) (Vis/skjul)
– en korrespondancetabel for variationsnavne. (download)
Kodningssekvens (Vis/Skjul)
CFTR-mutationerne
Der er indtil nu beskrevet næsten 1 700 CFTR-sekvensmutationer og -variationer (www.genet.sickkids.on.ca/cftr). Den mest almindelige mutation på verdensplan, der er ansvarlig for cystisk fibrose, er en deletion af tre baser, der koder for en phenylalaninrest ved position 508 kaldet p.Phe508del, som forringer CFTR-proteinets evne til at folde sig i det endoplasmatiske retikulum (ER) og derved øger den hurtige nedbrydning af proteinet under ER-processering. Den udgør ca. 70 % af CF-allelerne.
Heterolog ekspression af fuld længde p.Phe508del-CFTR-cDNA har vist, at dette mutantprotein kun opnår kerneglykosylering og ikke komplekse oligosakkaridkæder, og at det derfor ikke transporteres til celleoverfladen, hvilket er i overensstemmelse med observationen af fravær af mutantprotein i friske epitelier fra patientvæv. Blandt andre skadelige virkninger forringer p.Phe508del den korrekte foldning af CFTR og maskerer også konformationelt den diacidiske exitkode af tre rester i NBD1, som synes nødvendig for ER-eksport.
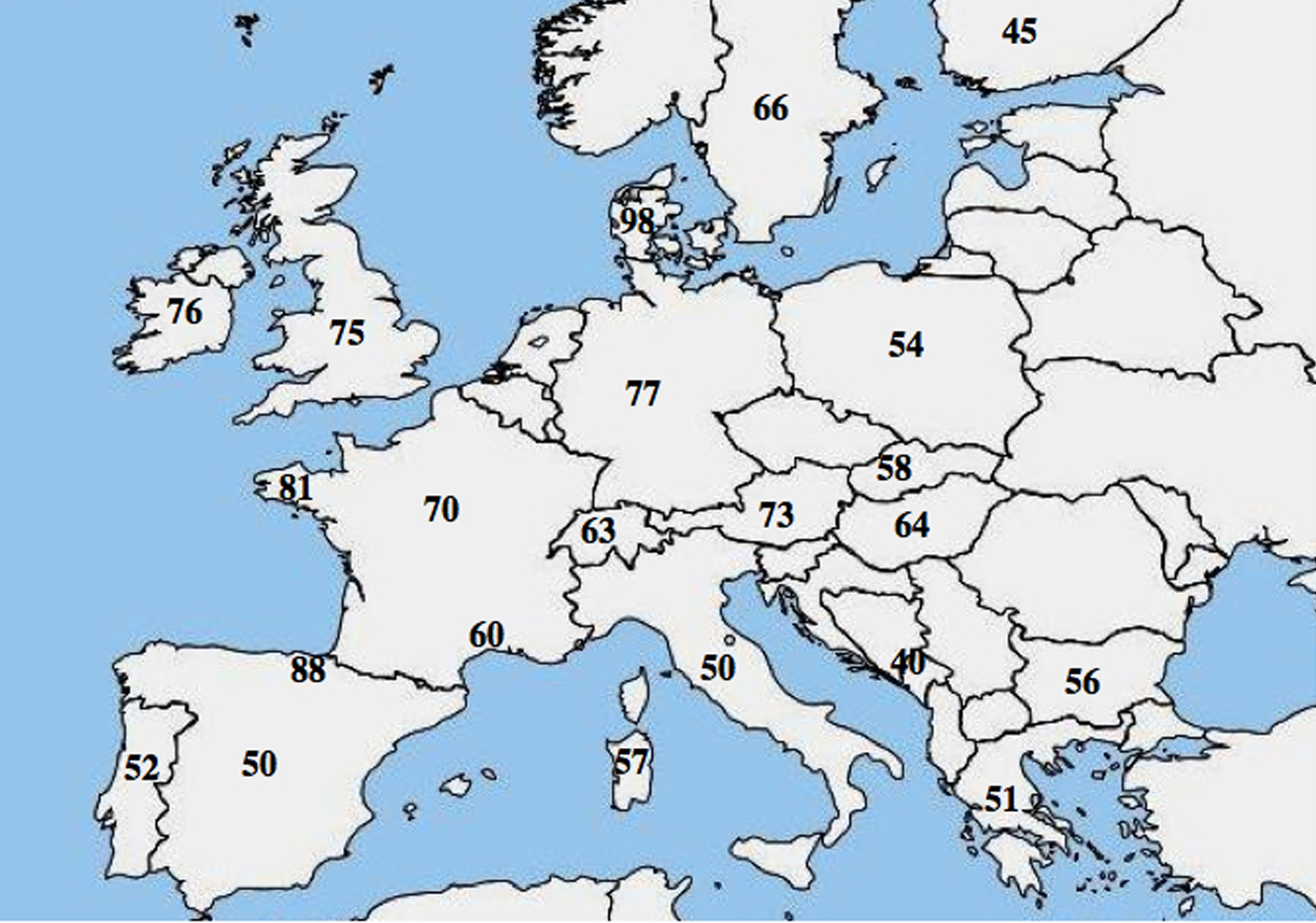
Der er blevet rapporteret om en molekylær allelisk heterogenitet i CF; adskillige undersøgelser fremhævede en markant forskel i hyppigheden af p.Phe508del blandt befolkninger og viste en faldende nordvestlig til sydøstlig gradient i Europa.
Klasser af CFTR-mutationer
Mutationer i CFTR-genet kan inddeles i seks klasser efter deres molekylære mekanismer og konsekvenser for forskellige aspekter af CFTR-biogenese, metabolisme og funktion.
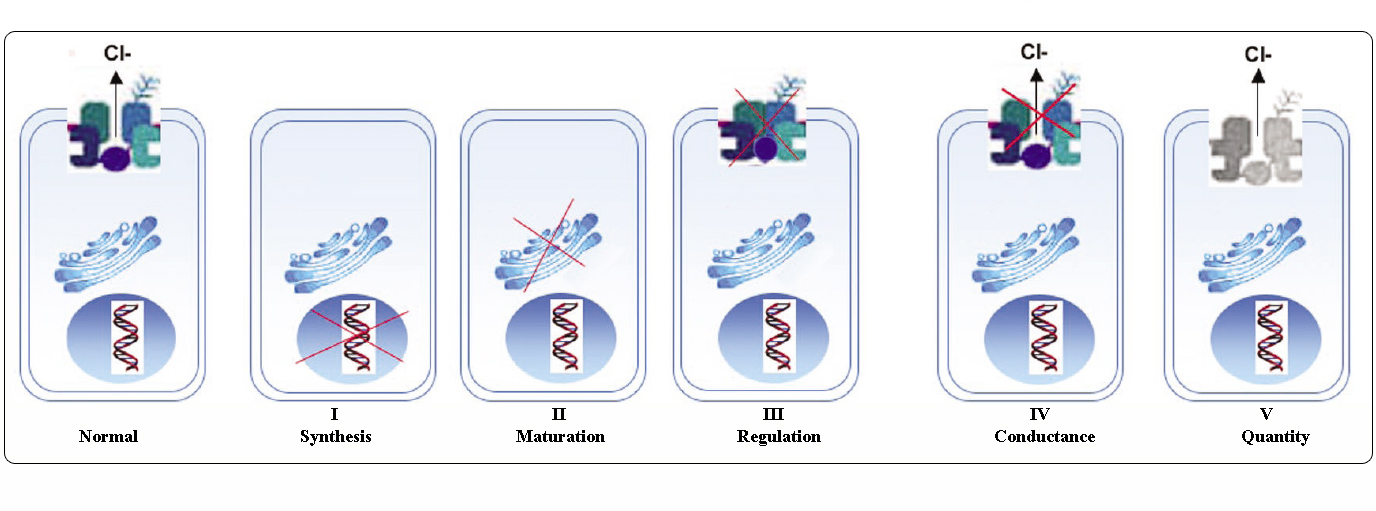
Adapteret fra Welsh og Smith (1993) og modificeret fra Claustres (RBM online, 2005)
Mutationer, der tilhører klasserne I-III og VI, giver kun lidt eller ingen funktionel CFTR ved den apikale membran og betragtes som “alvorlige” og fører normalt til en klassisk CF-fænotype med pancreasinsufficiens (CF-PI), selv om sværhedsgraden af lungesygdommen kan være varierende.
Mutationer, der tilhører klasse IV og V, bevarer en vis residual CFTR-aktivitet og giver en mildere fænotype. Hos patienter med mindst én “mild” CF-allel er CFTR-funktionen normalt tilstrækkelig til fordøjelsen, og lungesygdommen er mindre alvorlig. Personer med CFTR-mutanter, der giver anledning til ca. 10 % af det normale niveau af CFTR mRNA, kan kun have ét symptom, f.eks. CBAVD (congenital bilateral absence of vas deferens) hos mænd. Nogle personer, der er heterozygote for en CFTR-mutation, kan have en øget risiko for “CFTR-relaterede lidelser” (CFTR-RD) som f.eks. pancreatitis, bihulebetændelse eller allergisk bronchopulmonal aspergillose. I disse tilfælde kan CFTR-mutationer være en faktor for genetisk prædisponering, og genetiske eller miljømæssige modifikatorer kan have additive virkninger.
Gruppering af mutationer i forskellige klasser er nyttig til forståelse af dysfunktionens mekanisme, men mange sekvensvariationer er sjældne/unikke, og de tilgængelige data gør det ikke muligt at vurdere deres patogenicitet. Desuden kan en enkelt mutation forårsage mere end én type abnormitet eller give en variabel fænotype.
* Claustres M. 2005. Molekylær patologi af CFTR-lokussen i forbindelse med mandlig infertilitet. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Kompleksitet i en monogen sygdom. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identifikation af cystisk fibrosis-genet: genetisk analyse. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL m.fl. 1989. Identifikation af cystisk fibrosis-genet: kloning og karakterisering af komplementært DNA. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Fysisk lokalisering af to DNA-markører, der er tæt knyttet til cystisk fibrose-lokussen, ved hjælp af pulserende feltgelelektroforese. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. Molekylære mekanismer for CFTR-kloridkanal-dysfunktion ved cystisk fibrose. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Genomisk DNA-sekvens af genet for CFTR-genet (cystisk fibrose transmembrane conductance regulator). Genomics 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. CFTR-genvariant for patienter med medfødt fravær af vas deferens. Am J Hum Genet 57(4):958-60. PMID: 7573058.