Eine Frau in den 40ern stellte sich mit mehreren „Anfällen“ in der letzten Woche vor, die immer häufiger wurden. Sie fühlt sich schwindelig und ist dann nicht mehr ansprechbar. Die Anfälle dauern in der Regel etwa eine Minute und klingen dann ab. Sie hatte keine Schmerzen in der Brust. Als Kind hatte sie bereits Krampfanfälle, aber während dieser Anfälle ist keine Anfallsaktivität festzustellen.
Der Monitor zeigt Folgendes an:

Ventrikuläre Tachykardie, Frequenz ca. 220 Schläge pro Minute
Während dieses Rhythmus war sie wach, schützte ihre Atemwege, war blass, schweißgebadet und hatte kühle Extremitäten. Pulse waren vorhanden. Es gab keine Atemnot.
Ein 12-Kanal-EKG wurde aufgezeichnet:
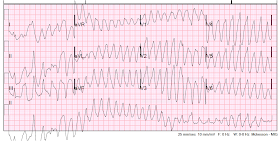
VT, und es handelt sich um eine „polymorphe“ VT, weil die Komplexe mehrere Morphologien aufweisen. Die Frequenz beträgt wiederum etwa 220. Polymorphe VT ist entweder eine Torsade (verbunden mit einem langen QT-Intervall) oder keine Torsade (QT nicht lang, oft aufgrund von Ischämie)
Auf dem Monitor wechselte sie spontan zu einem anderen Rhythmus, und es wurde ein weiteres 12-Kanal-Kardiogramm erstellt:

Es besteht eine Bigeminie. Den schmalen Komplexen gehen P-Wellen voraus. Das mit den Sinusschlägen verbundene QRS weist einen Rechtsschenkelblock (RBBB) auf und scheint kein sehr langes QT-Intervall zu haben (ich habe 400 ms QT geteilt durch die Quadratwurzel des vorangehenden R-R-Intervalls = 460 ms berechnet). Die dazwischen liegenden ventrikulären Komplexe (PVCs) sehen sehr bizarr aus und haben eine lange QT. Die PVCs haben eine große ST-Hebung (II, III, aVF) mit reziproker ST-Senkung (aVL, präkordial), was auf einen inferoposterioren STEMI hindeutet, aber dies ist eindeutig eine Mimik, da die dazwischen liegenden Sinuskomplexe keine ST-Hebung aufweisen.
Sie ist wieder tachykard geworden:
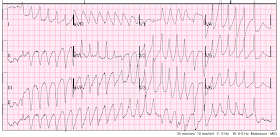
Polymorphe VT wieder.
Was sollte getan werden?
Erstens, um welche Art von VT handelt es sich? Polymorphe VT. Eine PMVT ist definiert als ein breitkomplexer ventrikulärer Rhythmus mit einer Frequenz von über 100 und einer sich schnell ändernden QRS-Achse und/oder Morphologie.
Polymorphe VT
Etiologie
Bei der polymorphen VT handelt es sich entweder um Torsades de pointe (verbunden mit einer langen QT auf dem 12-Kanal-Basis-EKG) oder um Nicht-Torsaden (in der Regel verbunden mit Ischämie oder anderen organischen Herzerkrankungen). Das Vorhandensein einer QT-Verlängerung auf dem 12-Kanal-Basis-EKG ist nicht immer offensichtlich. Torsade de pointes bedeutet „Verdrehung der Punkte“ und bezieht sich auf die Veränderung der Achse um eine isoelektrische Linie.
Die Morphologie der PMVT (d. h. das Vorhandensein einer Punktverdrehung) allein kann eine PMVT aufgrund einer langen QT (Torsaden) nicht von einer PMVT aufgrund anderer Ätiologien (Nicht-Torsaden) unterscheiden. Es ist praktisch unmöglich, eine pulslose PMVT von Kammerflimmern zu unterscheiden, und Studien haben gezeigt, dass die Mehrzahl der pulslosen Rhythmen, die wie Torsaden (mit „Verdrehung der Punkte“) aussehen, in Wirklichkeit Kammerflimmern sind. Diese Unterscheidung hat keine Auswirkungen auf die unmittelbare Behandlung (Defibrillation), aber sie hat wichtige Auswirkungen auf die Vermeidung weiterer Rhythmusstörungen.
Torsaden Ätiologien der PMVT
1. Erworben: meist durch Medikamente verursacht. Die Liste ist lang. Auch aufgrund von Elektrolytanomalien, insbesondere hypoK und hypoMg. Das korrigierte QT-Intervall (Bazett-Korrektur = QT geteilt durch die Quadratwurzel des vorangegangenen R-R-Intervalls in Millisekunden) ist normalerweise größer als 600 ms. Torsaden bei erworbener langer QT sind bei Bradykardie sehr viel wahrscheinlicher, da das QT-Intervall nach einer langen Pause noch länger ist. Daher werden Torsaden bei erworbener langer QT als „pausenabhängig“ bezeichnet: Wenn nach einer langen Pause ein Sinusschlag folgt (der ein längeres QT-Intervall erzeugt), ist es sehr viel wahrscheinlicher, dass eine frühe PVC („frühe Nachdepolarisation“, EAD) während der Repolarisation auftritt und Torsaden auslöst. Die übliche Abfolge ist: Sinusschlag, dann frühe PVC, dann eine lange Pause, weil die PVC früh war, was dann zu einer besonders langen QT führt, dann eine weitere PVC mit „R auf T“, die Torsaden auslöst.
2. Angeborenes, insbesondere angeborenes Long-QT-Syndrom. Eine instabile PMVT aufgrund eines angeborenen langen QT-Syndroms ist viel seltener. (In 26 Jahren EM und 125.000 Patienten habe ich noch nie einen Fall von Torsade PMVT aufgrund eines angeborenen langen QT-Syndroms gesehen). Zu den angeborenen Ursachen der Torsade gehört auch die „katecholaminerge PMVT“, bei der keine QT-Verlängerung auf dem 12-Kanal sichtbar ist, von der aber angenommen wird, dass sie eine ähnliche Ätiologie hat. Die präventive Therapie der angeborenen langen QT umfasst die Verwendung von Betablockern (wie bei der katecholaminergen PMVT, da in beiden Fällen die Betastimulation eine Torsade auslöst); dies ist ein Unterschied zur erworbenen langen QT, die mit Betastimulation (Isopreterenol) behandelt werden kann.
Ich habe keine Empfehlungen für den Einsatz von Betablockern zur akuten Behandlung von Torsaden in dieser Gruppe gefunden, aber es ist fast unmöglich zu sagen, ob Isoproterenol die Situation bei einem bestimmten Patienten mit angeborener langer QT verschlimmert oder verbessert (siehe Kommentar unseres Elektrophysiologen am Ende). Es scheint, dass die meisten symptomatischen Patienten mit angeborenem langem QT nach einer Synkope oder nach einer Reanimation mit Kammerflimmern auftreten und nur selten eine anhaltende Torsade oder Instabilität aufweisen. Daher besteht das Management dieser Patienten in erster Linie in der Erkennung und Verhinderung künftiger Synkopen und des plötzlichen Herztods, in der Regel mit einem implantierbaren Debribrillator zusätzlich zu Betablockern. Torsaden aufgrund einer angeborenen langen QT-Zeit können durch die Einnahme von QT-verlängernden Medikamenten ausgelöst werden.
Nicht-Torsade-Ätiologien von PMVT
Am häufigsten sind sie auf Ischämie zurückzuführen. Dabei handelt es sich fast immer um offene, schwere Ischämieepisoden mit Brustschmerzen und/oder eindeutigen ischämischen EKG-Anomalien. Sie können auch auf eine vorbestehende Kardiomyopathie zurückzuführen sein.
Behandlung von polymorphen VT
Die meisten Torsaden sind selbstlimitierend. Wenn sie sich nicht spontan umwandelt, muss sie defibrilliert werden, wenn der Patient instabil ist. Wenn sie sich umwandelt, ist es wahrscheinlich, dass sie erneut auftritt, und die Therapie zielt darauf ab, ein erneutes Auftreten zu verhindern.
Therapie von akuten Torsade-Episoden:
1. Kardioversion oder Defibrillation, wenn aktiv, besonders wenn instabil
2. Entfernung des auslösenden Agens bei erworbener langer QT
3. Korrektur von Hypokonzentration, auch auf leicht supranormale Werte
4. Verabreichung von 2-4 g MgSO4, auch wenn der Mg-Spiegel normal ist (eine Infusion von 3-10 mg pro Minute kann nützlich sein)
5. Nur wenn es sich um eine erworbene lange QT handelt: beta-adrenerge Stimulation mit Isoproterenol
6. Wenn dies nicht funktioniert, dann funktioniert fast immer eine überschießende Stimulation, normalerweise mit einer Frequenz von etwa 100, um Pausen zu vermeiden (transkutane Stimulation ist gut für eine vorübergehende Erleichterung als Überbrückung bis zur transvenösen Stimulation).
7. Lidocain kann auch von Nutzen sein, weil es die PVCs (frühe Nachdepolarisationen) unterdrücken kann, die eine Torsade auslösen, wenn sie auf der T-Welle auftreten.
8 Amiodaron ist von fragwürdigem Nutzen und möglicherweise schädlich. An sich verlängert es das QT-Intervall, ohne jedoch das Risiko von Torsaden wesentlich zu erhöhen
9. Geben Sie keine Betablocker, es sei denn, bei dem Patienten wurde eine angeborene lange QT-Zeit diagnostiziert. Genau das Gegenteil: Isoproterenol.
10. Wenn es angeboren ist, kann eine akute Betablockade angezeigt sein. Ich würde es zuerst mit Esmolol versuchen, da es abschaltbar ist. Es hat jedoch keine Beta-2-Blockade, und es ist mir unklar, ob dies wichtig und/oder notwendig ist. Wenn Esmolol nicht wirkt, sollte Propranolol intravenös verabreicht werden. Propranolol und Nadolol sind die besten Langzeit-Beta-Blocker bei angeborener langer QT, während das beta-1-selektive Metoprolol nicht sehr wirksam ist. Ob dies auf die Beta-Selektivität zurückzuführen ist, ist mir nicht klar.
Hier ist ein faszinierender Fall von angeborener langer QT mit Torsade. Die Rolle und Funktion der Betablockade bei kongenitaler langer QT wird ausführlich diskutiert.
Therapie der akuten PMVT ohne Torsade: Ähnlich wie bei monomorpher VT
1. Kardioversion oder Defibrillation, falls aktiv
2. Korrektur von Elektrolytstörungen, insbesondere hypoK oder hypoMg
3. Vorbeugung weiterer Episoden mit Lidocain oder Amiodaron, möglicherweise ein Betablocker wie Esmolol (den man bei erworbenen Torsaden mit langer QT vermeiden würde).
4. Anti-ischämische Therapien, bis hin zur Revaskularisation
5. Ein implantierbarer Kardioverter-Defibrillator kann auch bei erfolgreicher Revaskularisation erforderlich sein
Zurück zu unserem Fall
Eine Kardioversion ist nur dann angezeigt, wenn der Patient eine Torsade hat, und sie wirkt nur vorübergehend, da der Patient in den Rhythmus hinein- und wieder herausfährt. Die Verhinderung weiterer Episoden ist unerlässlich.
In diesem Fall sehen wir eine lange QT nur bei den PVCs, aber nicht bei den nativen RBBB-Schlägen. Oft sind die Torsaden „pausenabhängig“ und können nur in einem Komplex gesehen werden, der auf eine lange Pause folgt. Es treten keine Brustschmerzen auf, und die Sinus-EKG-Komplexe enthalten keine Hinweise auf eine Ischämie. Damit eine polymorphe VT auf eine Ischämie zurückzuführen ist, gibt es normalerweise eindeutige EKG-Befunde einer Ischämie. All dies deutet also auf Torsaden hin, ist aber nicht diagnostisch. Die Patientin nahm keine bekannten Medikamente ein, die das QT-Intervall verlängern. Sie hat lediglich eine Vorgeschichte mit Morbus Basedow und berichtet, dass sie keine Medikamente mehr einnimmt.
Wenn sie keine VT hat, ist ihr Blutdruck mit 190/80 erhöht, ihr Puls ist tastbar und liegt bei 90, die Sauerstoffsättigung bei 99.
Ein erster K-Wert war normal. Der Mg-Spiegel war zu diesem Zeitpunkt nicht bekannt.
Bei t = 12 wurden 2 g Magnesium intravenös verabreicht.
Bei t = 13 Minuten wurden 150 mg Amiodaron verabreicht
Bei t = 15 Minuten wurden 100 mg Lidocain verabreicht
Bei t = 30 Minuten trat keine Besserung ein, und es wurden Esmolol-Bolus und -Tropf verabreicht. Isoproterenol sollte gegeben werden.
Das Katheterlabor wurde aktiviert. Es wurden weitere 2 Gramm Magnesium intravenös verabreicht.
T = 64 Minuten: Der K-Wert liegt wieder bei 2,4 mEq/L (der Ausgangswert war falsch). Dies deutet noch stärker auf eine Torsade hin. KCl wird in die zentrale Leitung gegeben. Amiodaron-Tropf gestartet. Rektales Aspirin verabreicht.
t = 79 Minuten: Procainamid 1500 mg über 2 Minuten verabreicht. (Procainamid kann bei PMVT ohne Torsade nützlich sein, kann aber die QT verlängern und Torsaden instabiler machen. Außerdem ist es ein starkes negatives Inotropikum und könnte bei schlechter LV-Funktion gefährlich sein.)
t = 97 Minuten: Patient wird ins Katheterlabor gebracht:
Der Patient wurde ins Katheterlabor gebracht und ein Schrittmacher wurde platziert. Der Rhythmus wurde mit Overdrive-Schrittmachern eingefangen und dann verlangsamt. Es bestand eine diffuse Koronarerkrankung, aber keine kausale Läsion (kein akutes Koronarsyndrom). Der Troponinwert war negativ.
Es wurde festgestellt, dass der Patient Methadon einnahm und methadongiftig war. Es ist eines der vielen Medikamente, die eine lange QT-Zeit verursachen. Es handelte sich um Torsade de Pointes aufgrund einer langen QT-Zeit durch Methadon und Hypokaliämie.
Hier ist das 12-Kanal-Kabel während der Stimulation:
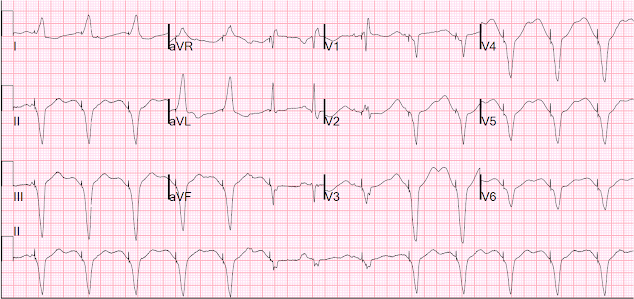
Während der Stimulation
Hier ist ein weiteres mit ausgeschaltetem Schrittmacher, am nächsten Tag:
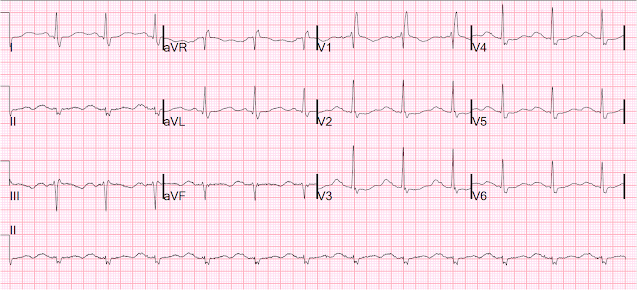
Sinus mit RBBB und sehr langer QT
Und dann am vierten Tag:
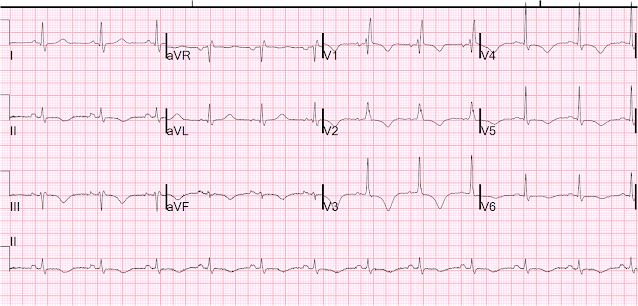
T-Wellen-Inversionen, QT immer noch lang
Aufgrund der außerordentlich langen Halbwertszeit von Methadon dauerte es Tage, bis sich die QT verkürzte. Letztendlich ging es dem Patienten gut.
Bemerkung zum Einsatz von Isoproterenol bei angeborener langer QT:
Ich habe unserem Elektrophysiologen (Rehan Karim) folgende Frage gestellt:
„Ich möchte sicher sein, dass ich das richtig verstanden habe:
Bei angeborener langer QT ist eine chronische Betablockade indiziert, und bei einem Patienten mit rezidivierender VT, der sich im Krankenhaus befindet, würde man NICHT Isoproterenol geben.
Nur bei einer erworbenen langen QT, die „pausenabhängig“ ist und sich mit einer schnelleren Herzfrequenz verbessert, ist eine Betablockade NICHT angezeigt. Vielmehr ist Isoproterenol indiziert (und Übersteuerungsstimulation).“
Können Sie mich diesbezüglich korrigieren?“
Mit anderen Worten:
Hier ist seine Antwort:
„Ich wünschte, die Dinge wären klar genug, um alles in ein algorithmisches Format zu bringen… aber das ist leider nicht der Fall.“
Es hängt wirklich davon ab, mit welcher Art von angeborener langer QT man es zu tun hat, und selbst wenn man es weiß – die Dinge sind nicht so einfach.
Hier ein Beispiel:
Bei der angeborenen langen QT vom Typ 3 handelt es sich um eine Natriumkanalmutation (SCN5A) – das ist das gleiche Gen wie beim Brugada-Syndrom.
Bei der langen QT vom Typ 3 treten pausenabhängige Torsades auf. Dennoch werden in dieser Situation Betablocker eingesetzt (nachdem natürlich eine Vorhofstimulation eingeleitet wurde, um eine Bradykardie zu verhindern) – in erster Linie wegen der Wirkung der Betablocker auf die Verringerung der QT-Dispersion.
Es wird noch komplizierter… Natriumkanalblocker der Klasse I-B (Mexiletin) werden manchmal bei LQT-3 eingesetzt.
Meines Wissens sind die Betablocker bei LQT-3 wahrscheinlich nicht so wirksam wie bei anderen Formen – aber sie werden dennoch eingesetzt.
Deshalb glaube ich nicht, dass es möglich ist, mit dem Wissen, das wir derzeit haben, eine Ja/Nein-Antwort zu geben – die Dinge müssen aus der klinischen Perspektive betrachtet werden.
Wenn jemand eindeutig an pausenabhängigen Torsaden leidet und nicht mit dem Herzschrittmacher behandelt wird, würde ich KEINEN Betablocker einsetzen, sondern versuchen, die Herzfrequenz mit Isoproterenol zu erhöhen.
Dr. Karim fügte hinzu:
Dies ist eines der verwirrendsten Themen – tatsächlich sind die meisten genetischen Arrhythmien / Chanellopathien klinisch ziemlich schwierig zu handhaben. Wir sind so sehr daran gewöhnt, Daten von Tausenden von Patienten in kardiologischen Studien zu sehen – und dann kommen wir in solche Situationen, in denen die Krankheitslast nicht groß genug ist, um uns andere Daten als Register von großen klinischen Zentren zu liefern.