 |
A cisztás fibrózis transzmembrán konduktancia szabályozó gén (CFTR, OMIM #602421) az ATP-kötő kazetták C alcsaládjának (ABCC7) tagja. Az 1989-ben felfedezett gén (Kerem et al, 1989; Riordan et al, 1989; Rommens et al, 1989) a 7. kromoszóma hosszú karján (7q31-q32 sáv) (pozíció 117,119,358-117,308,719) 188,702 bp-t tesz ki, és 27 exonból áll, amelyek kezdetben az 1-24-es számozásúak, a 6., 14. és 17. exonok a és b alosztállyal (Zielenski et al., 1991). Az érett transzkript 6 129 nukleotid hosszú, beleértve egy 4 440 kódoló bázisból álló nyílt olvasási keretet (referencia szekvencia: NM_000492). Ensembl Gene ID: ENSG00000001626 Chromosome 7 ( NCBI, vagy HGNC) |
Nómenklatúra
A UMD-CFTR a Human Genome Variation Society (www.hgvs.org/mutnomen/) által javasolt ajánlott nómenklatúra irányelveket használja, pl., cDNS-alapú számozás, ahol az ATG transzlációs iniciációs kódon A-ja a +1, az exonok pedig 1-től 27-ig vannak számozva.
Az UMD-CFTR-ben a kódoló referencia szekvencia az NM_000492, kivéve a c.1408A-t a G helyett, és a referencia fehérjeszekvencia az NP_000483, kivéve a p.Met470-t a p.Val470 helyett.
A cisztás fibrózis mutációs adatbázis konzorciumi számozásához (www.genet.sickkids.on.ca/cftr/) szokott felhasználók kényelmének érdekében két letölthető fájlt biztosítunk:
– az exonok és nukleotidok megfelelő számozása: közös számozás (fehérrel az exonoknál és feketével a nukleotidoknál) és ajánlott számozás (pirossal). Az első és utolsó nukleotidok minden egyes exon esetében fel vannak tüntetve. (Letöltés) (Megjelenítés/elrejtés)
– a variációs nevek megfelelési táblázata. (letöltés)
Kódoló szekvencia (Show/Hide)
A CFTR mutációk
Már csaknem 1700 CFTR szekvencia mutációt és variációt írtak le eddig (www.genet.sickkids.on.ca/cftr). A világszerte leggyakoribb mutáció, amely a cisztás fibrózisért felelős, az 508-as pozícióban lévő fenilalanin-maradékot kódoló három bázis deléciója, a p.Phe508del, amely rontja a CFTR fehérjének az endoplazmatikus retikulumban (ER) történő hajtogatási képességét, ezáltal fokozza a fehérje gyors lebomlását az ER-feldolgozás során. Ez a CF allélok mintegy 70%-át teszi ki.
A teljes hosszúságú p.Phe508del-CFTR-cDNS heterológ expressziója kimutatta, hogy ez a mutáns fehérje csak magglikozilációt kap, komplex oligoszacharidláncokat nem, és mint ilyen, nem tud a sejtfelszínre transzportálni, összhangban azzal a megfigyeléssel, hogy a beteg szövetekből származó friss epithéliumokban a mutáns fehérje hiányzik. Egyéb káros hatások mellett a p.Phe508del rontja a CFTR helyes hajtogatódását, és konformációs szempontból elrejti az NBD1 három maradékának diaszidikus kilépési kódját is, amely szükségesnek tűnik az ER-exporthoz.
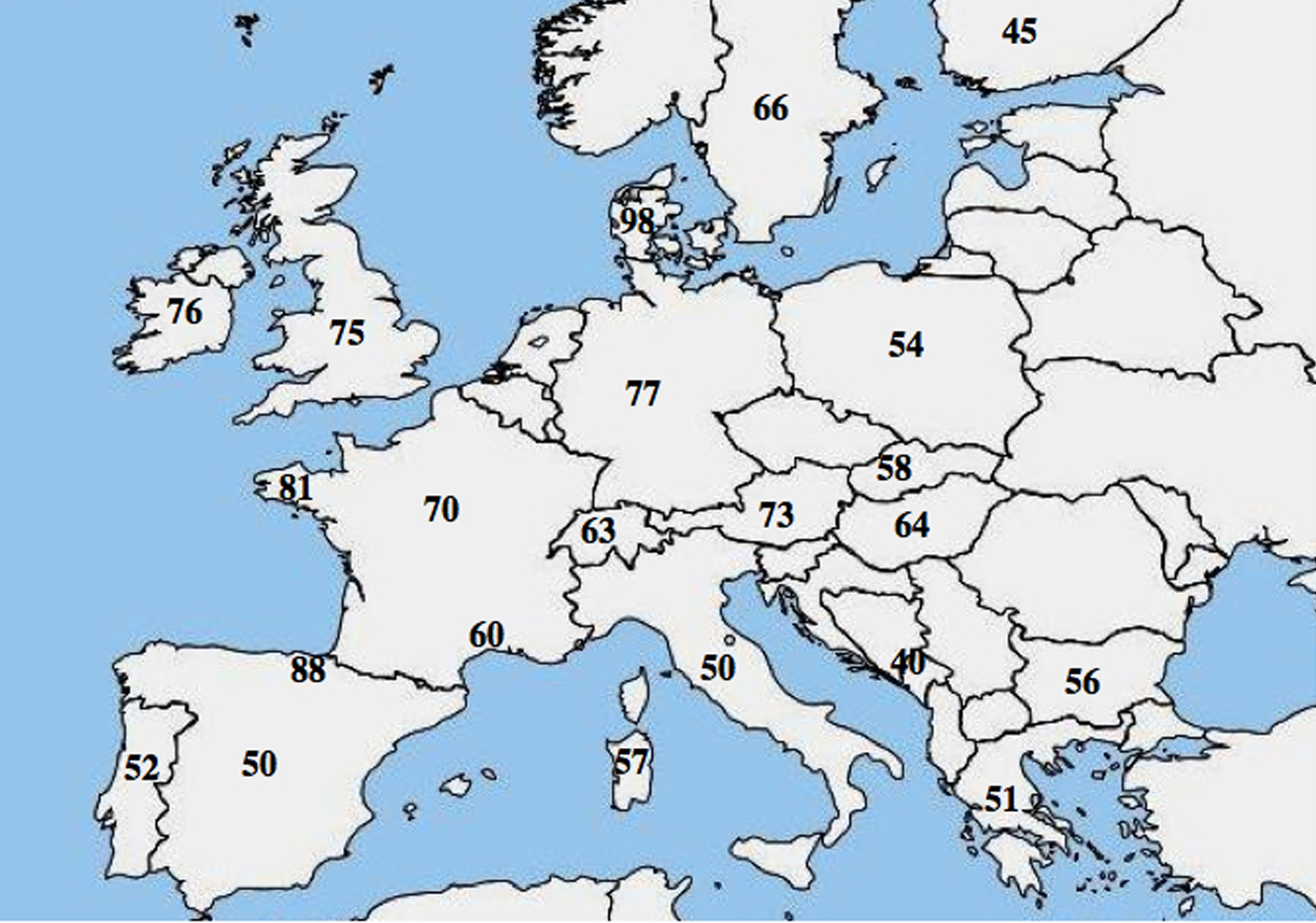
A CF-ben molekuláris allelikus heterogenitásról számoltak be; számos tanulmány kiemelte a p.Phe508del gyakoriságának jelentős különbségét a populációk között, és Európában csökkenő északnyugat-délkeleti gradienst mutatott ki.
A CFTR-mutációk osztályai
A CFTR-gén mutációi hat osztályba sorolhatók molekuláris mechanizmusuk és a CFTR biogenezis, metabolizmus és funkció különböző aspektusaira gyakorolt következményeik alapján.
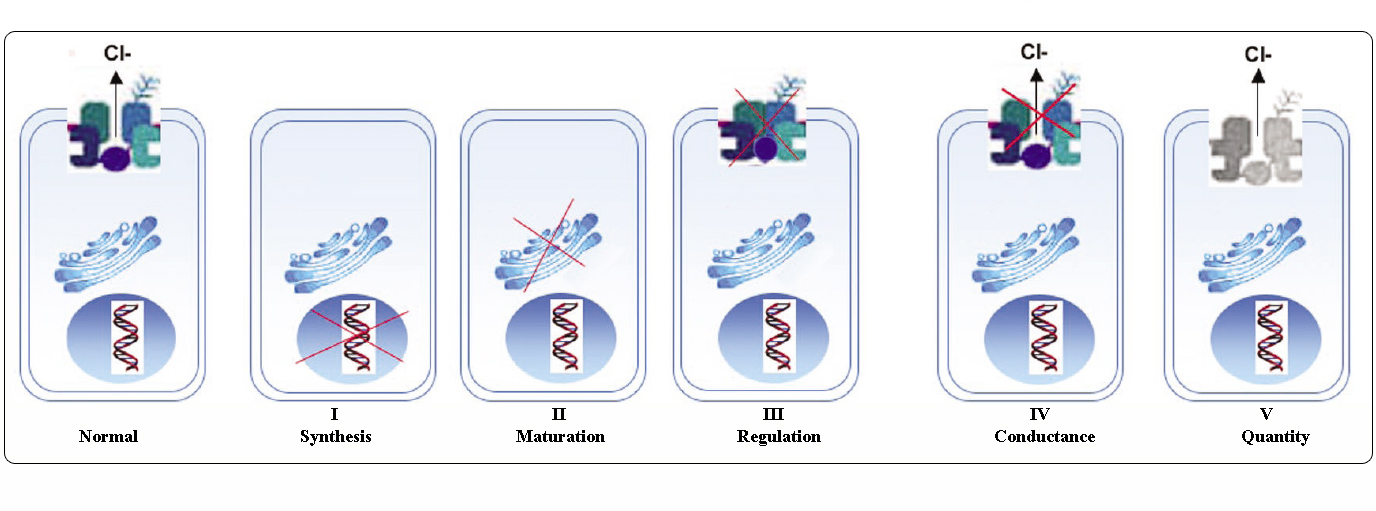
Welsh és Smith (1993) alapján átvéve és Claustres (RBM online, 2005) alapján módosítva
Az I-III. és VI. osztályba tartozó mutációk kevés vagy egyáltalán nem funkcionális CFTR-t kölcsönöznek az apikális membránon, “súlyosnak” tekinthetők és általában klasszikus CF fenotípushoz vezetnek hasnyálmirigy-elégtelenséggel (CF-PI), bár a tüdőbetegség súlyossága változó lehet.
A IV. és V. osztályba tartozó mutációk megtartják a CFTR némi maradék aktivitását, és enyhébb fenotípust eredményeznek. A legalább egy “enyhe” CF-alléllel rendelkező betegeknél a CFTR-funkció általában elegendő az emésztéshez, és a tüdőbetegség kevésbé súlyos. Az olyan CFTR-mutánsokkal rendelkező egyéneknél, amelyek a CFTR mRNS normális szintjének körülbelül 10%-át adják, előfordulhat, hogy csak egy tünet jelentkezik, mint például a CBAVD (veleszületett kétoldali vas deferens hiánya) férfiaknál. Egyes CFTR-mutációra heterozigóta egyéneknél megnövekedhet a “CFTR-rel kapcsolatos rendellenességek” (CFTR-RD), például hasnyálmirigy-gyulladás, arcüreggyulladás vagy allergiás bronchopulmonális aspergillózis kockázata. Ezekben az esetekben a CFTR-mutáció a genetikai hajlam egyik tényezője lehet, és a genetikai vagy környezeti módosítók additív hatást gyakorolhatnak.
A mutációk különböző osztályokba sorolása hasznos a működési zavar mechanizmusának megértéséhez, azonban sok szekvencia-variáció ritka/egyedi, és a rendelkezésre álló adatok nem teszik lehetővé patogenitásuk értékelését. Ráadásul egyetlen mutáció többféle rendellenességet is okozhat, vagy változó fenotípust eredményezhet.
* Claustres M. 2005. A CFTR-lókusz molekuláris patológiája a férfi meddőségben. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Komplexitás egy monogén betegségben. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. A cisztás fibrózis génjének azonosítása: genetikai elemzés. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL és mások. 1989. A cisztás fibrózis génjének azonosítása: a komplementer DNS klónozása és jellemzése. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Két, a cisztás fibrózis lókuszához szorosan kapcsolódó DNS marker fizikai lokalizációja pulzálómezős gélelektroforézissel. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. A CFTR kloridcsatorna diszfunkciójának molekuláris mechanizmusai cisztás fibrózisban. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. A cisztás fibrózis transzmembrán konduktancia szabályozó (CFTR) gén genomiális DNS szekvenciája. Genomics 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. CFTR génváltozat a veleszületett vas deferens hiányban szenvedő betegeknél. Am J Hum Genet 57(4):958-60. PMID: 7573058.