 |
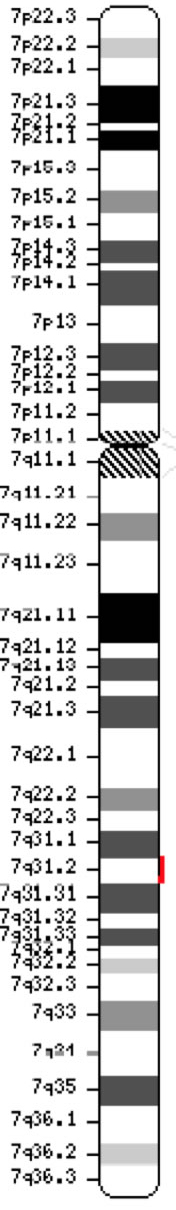
Gen regulatora transmembranowego przewodnictwa w mukowiscydozie (CFTR, OMIM #602421) jest członkiem podrodziny C kaset wiążących ATP (ABCC7). Gen ten, odkryty w 1989 roku (Kerem i wsp., 1989; Riordan i wsp., 1989; Rommens i wsp., 1989), rozciąga się na 188 702 bp na długim ramieniu chromosomu 7 (pasmo 7q31-q32) (pozycja 117 119 358-117 308 719) i składa się z 27 eksonów oznaczonych początkowo numerami 1-24 z podrozdziałami a i b dla eksonów 6, 14 i 17 (Zielenski i wsp., 1991). Dojrzały transkrypt ma długość 6 129 nukleotydów, w tym otwartą ramkę odczytu zawierającą 4 440 zasad kodujących (sekwencja referencyjna: NM_000492). Ensembl Gene ID: ENSG00000001626 Chromosom 7 ( NCBI, lub HGNC) |
Nomenklatura
W UMD-CFTR zastosowano zalecane wytyczne nomenklatury zaproponowane przez Human Genome Variation Society (www.hgvs.org/mutnomen/) np, numeracja oparta na cDNA z kodonem A inicjacji translacji ATG przy +1 i eksonami ponumerowanymi od 1 do 27.
W UMD-CFTR kodującą sekwencją referencyjną jest NM_000492 z wyjątkiem c.1408A zamiast G, a referencyjną sekwencją białkową jest NP_000483 z wyjątkiem p.Met470 zamiast p.Val470.
Dla wygody użytkowników, którzy są przyzwyczajeni do numeracji konsorcjum bazy danych mutacji mukowiscydozy (www.genet.sickkids.on.ca/cftr/), dostarczane są dwa pliki do pobrania:
– numeracja korespondencyjna dla eksonów i nukleotydów: numeracja wspólna (w kolorze białym dla eksonów i w kolorze czarnym dla nukleotydów) oraz numeracja zalecana (w kolorze czerwonym). Dla każdego eksonu podano pierwszy i ostatni nukleotyd. (pobierz) (Pokaż/Ukryj)
– tabela korespondencji dla nazw wariantów. (download)
Sekwencja kodująca (Show/Hide)
Mutacje CFTR
Do tej pory opisano prawie 1700 mutacji i wariantów sekwencji CFTR (www.genet.sickkids.on.ca/cftr). Najczęstszą mutacją na świecie odpowiedzialną za mukowiscydozę jest delecja trzech zasad kodujących resztę fenyloalaninową w pozycji 508 o nazwie p.Phe508del, która upośledza zdolność białka CFTR do składania się w retikulum endoplazmatycznym (ER), zwiększając tym samym szybką degradację białka podczas przetwarzania ER. Stanowi ona około 70% alleli CF.
Heterologiczna ekspresja pełnometrażowego p.Phe508del-CFTR-cDNA wykazała, że to zmutowane białko nabywa tylko rdzeniową glikozylację, a nie złożone łańcuchy oligosacharydowe i jako takie nie jest transportowane na powierzchnię komórki, zgodnie z obserwacją braku zmutowanego białka w świeżym nabłonku z tkanek pacjenta. Wśród innych szkodliwych efektów, p.Phe508del upośledza prawidłowe składanie CFTR, a także konformacyjnie maskuje diacydowy kod wyjścia trzech reszt w NBD1, który wydaje się niezbędny do eksportu ER.
Zgłaszano molekularną heterogenność alleliczną w CF; liczne badania podkreśliły wyraźną różnicę w częstości występowania p.Phe508del wśród populacji i wykazały zmniejszający się gradient z północnego zachodu na południowy wschód w Europie.
Klasy mutacji CFTR
Mutacje w genie CFTR można podzielić na sześć klas w zależności od ich mechanizmów molekularnych i konsekwencji dla różnych aspektów biogenezy, metabolizmu i funkcji CFTR.
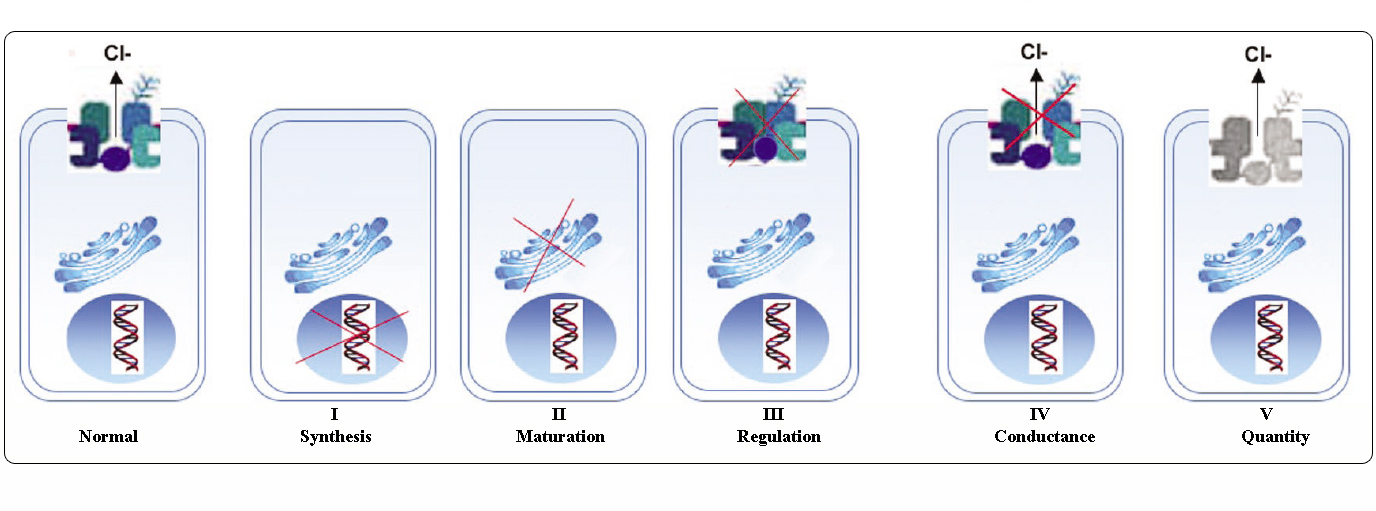
Zaadaptowane z Welsh i Smith (1993) i zmodyfikowane z Claustres (RBM online, 2005)
Mutacje należące do klas I-III i VI w niewielkim stopniu lub wcale nie dają funkcjonalnego CFTR w błonie apikalnej i są uważane za „ciężkie” i zwykle prowadzą do klasycznego fenotypu CF z niewydolnością trzustki (CF-PI), chociaż ciężkość choroby płuc może być zmienna.
Mutacje należące do klas IV i V zachowują pewną resztkową aktywność CFTR i nadają łagodniejszy fenotyp. U pacjentów z co najmniej jednym „łagodnym” allelem CF, funkcja CFTR jest zazwyczaj wystarczająca do trawienia, a choroba płuc jest mniej poważna. Osoby z mutacjami CFTR, które powodują powstanie około 10% normalnego poziomu mRNA CFTR, mogą prezentować tylko jeden objaw, taki jak CBAVD (wrodzony obustronny brak nasieniowodów) u mężczyzn. Niektóre osoby heterozygotyczne dla mutacji CFTR mogą być narażone na zwiększone ryzyko wystąpienia „zaburzeń związanych z CFTR” (CFTR-RD), takich jak zapalenie trzustki, zapalenie zatok lub alergiczna aspergiloza oskrzelowo-płucna. W tych przypadkach mutacje CFTR mogą być czynnikiem predyspozycji genetycznej, a modyfikatory genetyczne lub środowiskowe mogą odgrywać rolę addytywną.
Grupowanie mutacji w różne klasy jest przydatne do zrozumienia mechanizmu dysfunkcji, jednak wiele wariantów sekwencji jest rzadkich/unikalnych i dostępne dane nie pozwalają na ocenę ich patogenności. Co więcej, pojedyncza mutacja może powodować więcej niż jeden typ nieprawidłowości lub może nadawać zmienny fenotyp.
* Claustres M. 2005. Molekularna patologia locus CFTR w niepłodności męskiej. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Complexity in a monogenic disease. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identification of the cystic fibrosis gene: genetic analysis. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL i inni. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Physical localization of two DNA markers closely linked to the cystic fibrosis locus by pulsed-field gel electrophoresis. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73(7):1251-4. PMID: 7686820.
* Zieleński J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 10(1):214-28. PMID: 1710598.
* Zieleński J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. CFTR gene variant for patients with congenital absence of vas deferens. Am J Hum Genet 57(4):958-60. PMID: 7573058.
.