 |
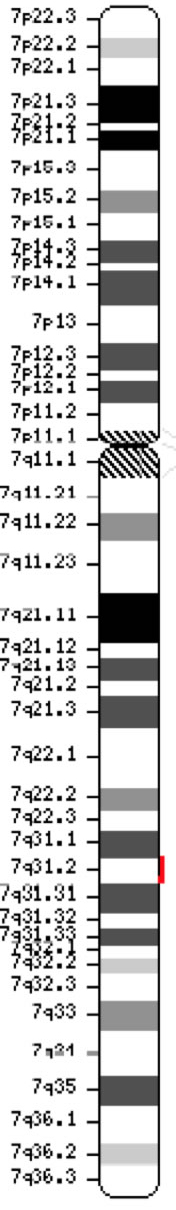
Il gene regolatore della conduttanza transmembrana della fibrosi cistica (CFTR, OMIM #602421) è un membro della sottofamiglia C della cassetta di legame ATP (ABCC7). Il gene, scoperto nel 1989 (Kerem et al, 1989; Riordan et al, 1989; Rommens et al, 1989), si estende per 188.702 bp sul braccio lungo del cromosoma 7 (banda 7q31-q32) (posizione 117.119.358-117.308.719) ed è composto da 27 esoni inizialmente numerati 1-24 con suddivisioni a e b per gli esoni 6, 14 e 17 (Zielenski et al., 1991). Il trascritto maturo è lungo 6.129 nucleotidi e comprende un open reading frame di 4.440 basi codificanti (sequenza di riferimento: NM_000492). Ensembl Gene ID: ENSG00000001626 Chromosome 7 ( NCBI, o HGNC) |
Nomenclatura
L’UMD-CFTR usa le linee guida raccomandate per la nomenclatura proposte dalla Human Genome Variation Society (www.hgvs.org/mutnomen/) ad es, numerazione basata sul cDNA con la A del codone di inizio traslazione ATG a +1 e gli esoni numerati da 1 a 27.
Nell’UMD-CFTR la sequenza di riferimento codificante è NM_000492 con l’eccezione di c.1408A invece di G, e la sequenza proteica di riferimento è NP_000483 con l’eccezione di p.Met470 invece di p.Val470.
Per comodità degli utenti che sono abituati alla numerazione del consorzio del database delle mutazioni della fibrosi cistica (www.genet.sickkids.on.ca/cftr/) sono forniti due file da scaricare:
– una numerazione di corrispondenza per esoni e nucleotidi: numerazione comune (in bianco per gli esoni e in nero per i nucleotidi) e numerazione raccomandata (in rosso). Il primo e l’ultimo nucleotide sono indicati per ogni esone. (download) (Show/Hide)
– una tabella di corrispondenza per i nomi delle variazioni. (download)
Seguenza codificante (Mostra/Nascondi)
Le mutazioni CFTR
Sono state descritte finora quasi 1.700 mutazioni e variazioni della sequenza CFTR (www.genet.sickkids.on.ca/cftr). La mutazione più comune a livello mondiale responsabile della fibrosi cistica è una delezione di tre basi che codifica un residuo di fenilalanina in posizione 508 chiamata p.Phe508del, che compromette la capacità della proteina CFTR di ripiegarsi nel reticolo endoplasmatico (ER), aumentando così la rapida degradazione della proteina durante il processamento dell’ER. Rappresenta circa il 70% degli alleli CF.
L’espressione eterologa di p.Phe508del-CFTR-cDNA ha dimostrato che questa proteina mutante acquisisce solo la glicosilazione del nucleo e non catene oligosaccaridiche complesse e, come tale, non riesce ad essere trasportata alla superficie cellulare, in accordo con l’osservazione dell’assenza della proteina mutante in epiteli freschi di tessuti di pazienti. Tra gli altri effetti dannosi, p.Phe508del compromette il corretto ripiegamento di CFTR e inoltre maschera conformazionalmente il codice diacidico di uscita di tre residui nel NBD1 che sembra necessario per l’esportazione ER.
È stata riportata un’eterogeneità molecolare allelica nella FC; numerosi studi hanno evidenziato una marcata differenza nella frequenza di p.Phe508del tra le popolazioni e hanno mostrato un gradiente decrescente da nord ovest a sud est in Europa.
Classi di mutazioni CFTR
Le mutazioni nel gene CFTR possono essere classificate in sei classi in base ai loro meccanismi molecolari e alle conseguenze su diversi aspetti della biogenesi, del metabolismo e della funzione di CFTR.
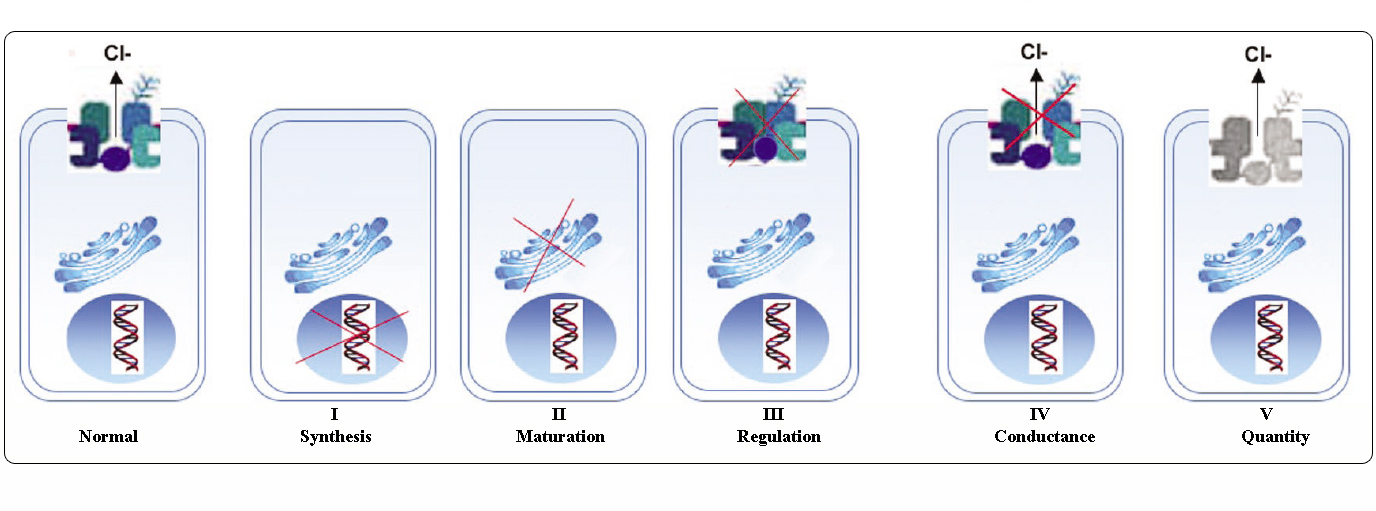
Adattato da Welsh e Smith (1993) e modificato da Claustres (RBM online, 2005)
Le mutazioni appartenenti alle classi I-III e VI conferiscono poca o nessuna CFTR funzionale alla membrana apicale e sono considerate “gravi” e solitamente portano ad un fenotipo classico di CF con insufficienza pancreatica (CF-PI) anche se la gravità della malattia polmonare può essere variabile.
Le mutazioni appartenenti alle classi IV e V mantengono una certa attività CFTR residua e conferiscono un fenotipo più lieve. Nei pazienti con almeno un allele CF “lieve”, la funzione CFTR è solitamente sufficiente per la digestione e la malattia polmonare è meno grave. Gli individui con mutanti CFTR che danno origine a circa il 10% del livello normale di mRNA CFTR possono presentare un solo sintomo, come la CBAVD (assenza bilaterale congenita dei vasi deferenti) nei maschi. Alcuni individui eterozigoti per una mutazione CFTR potrebbero essere a maggior rischio di “disturbi legati alla CFTR” (CFTR-RD) come pancreatite, sinusite o aspergillosi broncopolmonare allergica. In questi casi, le mutazioni CFTR potrebbero essere un fattore di predisposizione genetica e i modificatori genetici o ambientali potrebbero giocare effetti additivi.
Raggruppare le mutazioni in diverse classi è utile per comprendere il meccanismo della disfunzione, tuttavia molte variazioni di sequenza sono rare/uniche e i dati disponibili non permettono di valutare la loro patogenicità. Inoltre, una singola mutazione può causare più di un tipo di anomalia o può conferire un fenotipo variabile.
* Claustres M. 2005. Patologia molecolare del locus CFTR nell’infertilità maschile. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Complessità in una malattia monogenica. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identificazione del gene della fibrosi cistica: analisi genetica. Scienza 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL e altri. 1989. Identificazione del gene della fibrosi cistica: clonazione e caratterizzazione del DNA complementare. Scienza 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Localizzazione fisica di due marcatori del DNA strettamente legati al locus della fibrosi cistica mediante elettroforesi su gel a campo pulsato. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Gallese MJ, Smith AE. 1993. Meccanismi molecolari della disfunzione del canale del cloruro CFTR nella fibrosi cistica. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Sequenza del DNA genomico del gene del regolatore di conduttanza transmembrana della fibrosi cistica (CFTR). Genomics 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. Variante del gene CFTR per i pazienti con assenza congenita dei vasi deferenti. Am J Hum Genet 57(4):958-60. PMID: 7573058.