 |
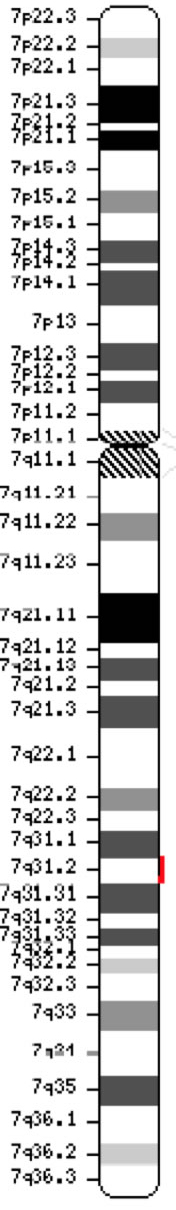
Gena regulatorului de conductanță transmembranară a fibrozei chistice (CFTR, OMIM #602421) este membră a subfamiliei C (ABCC7) a casetelor de legare a ATP (ATP-binding cassette sub-family C). Gena, descoperită în 1989 (Kerem et al, 1989; Riordan et al, 1989; Rommens et al, 1989), se întinde pe 188.702 pb pe brațul lung al cromozomului 7 (banda 7q31-q32) (poziția 117.119.358-117.308.719) și este compusă din 27 de exoni numerotați inițial 1-24 cu subdiviziunile a și b pentru exonii 6, 14 și 17 (Zielenski et al., 1991). Transcriptul matur are o lungime de 6 129 de nucleotide, incluzând un cadru de lectură deschis de 4 440 de baze codificatoare (Secvența de referință: NM_000492). Ensembl Gene ID: ENSG00000001626 Cromosomul 7 ( NCBI, sau HGNC) |
Nomenclatură
UDM-CFTR utilizează liniile directoare de nomenclatură recomandate propuse de Human Genome Variation Society (www.hgvs.org/mutnomen/), de ex, numerotarea bazată pe ADNc, cu codonul de inițiere a translației A de ATG la +1 și exonii numerotați de la 1 la 27.
În UMD-CFTR secvența de referință codificatoare este NM_000492 cu excepția c.1408A în loc de G, iar secvența proteică de referință este NP_000483 cu excepția p.Met470 în loc de p.Val470.
Pentru comoditatea utilizatorilor care sunt obișnuiți cu numerotarea consorțiului bazei de date a mutațiilor din fibroza chistică (www.genet.sickkids.on.ca/cftr/) sunt furnizate două fișiere care pot fi descărcate:
– o numerotare de corespondență pentru exoni și nucleotide: numerotare comună (în alb pentru exoni și în negru pentru nucleotide) și numerotare recomandată (în roșu). Prima și ultima nucleotidă sunt indicate pentru fiecare exon. (descărcați) (Afișați/ Ascundeți)
– un tabel de corespondență pentru numele variațiilor. (download)
Secvența de codificare (Show/Hide)
Mutațiile CFTR
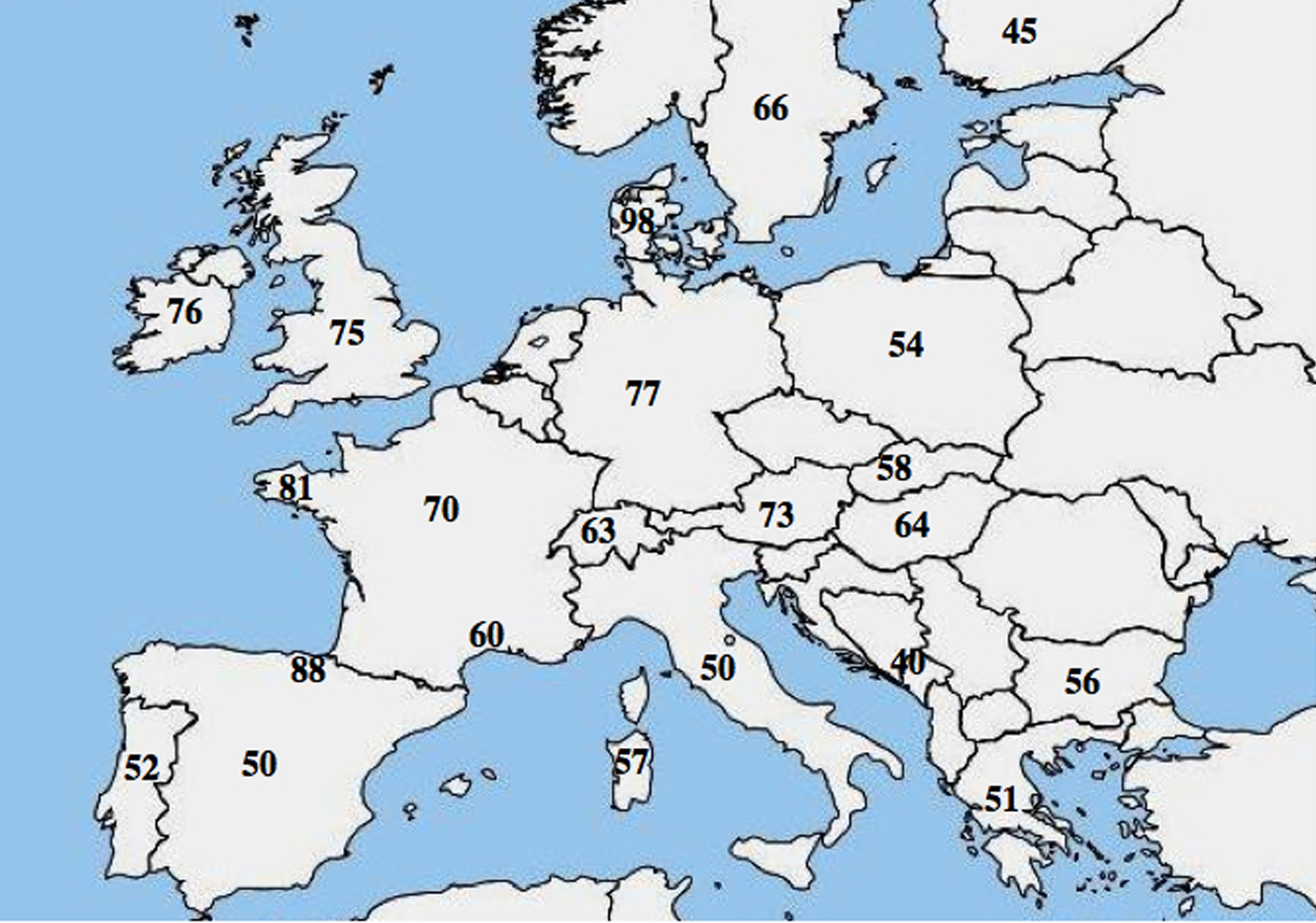
Aproape 1.700 de mutații și variații ale secvenței CFTR au fost descrise până în prezent (www.genet.sickkids.on.ca/cftr). Cea mai frecventă mutație la nivel mondial responsabilă de fibroza chistică este o deleție de trei baze care codifică un reziduu de fenilalanină în poziția 508 numită p.Phe508del, care afectează capacitatea proteinei CFTR de a se plia în reticulul endoplasmatic (ER), sporind astfel degradarea rapidă a proteinei în timpul procesării ER. Aceasta reprezintă aproximativ 70% din alelele FC.
Expresia heterologă a ADN-cFTR p.Phe508del-CFTR de lungime completă a demonstrat că această proteină mutantă dobândește doar glicozilarea nucleului și nu lanțuri oligozaharidice complexe și, ca atare, nu reușește să fie transportată la suprafața celulară, în conformitate cu observația absenței proteinei mutante în epiteliile proaspete din țesuturile pacienților. Printre alte efecte dăunătoare, p.Phe508del afectează plierea corectă a CFTR și, de asemenea, maschează conformațional codul de ieșire diacidic a trei reziduuri din NBD1, care pare a fi necesar pentru exportul ER.
A fost raportată o eterogenitate moleculară alelică în FC; numeroase studii au evidențiat o diferență marcantă a frecvenței p.Phe508del între populații și au arătat un gradient descrescător de la nord-vest la sud-est în Europa.
Classe de mutații CFTR
Mutațiile din gena CFTR pot fi clasificate în șase clase în funcție de mecanismele lor moleculare și de consecințele asupra diferitelor aspecte ale biogenezei, metabolismului și funcției CFTR.
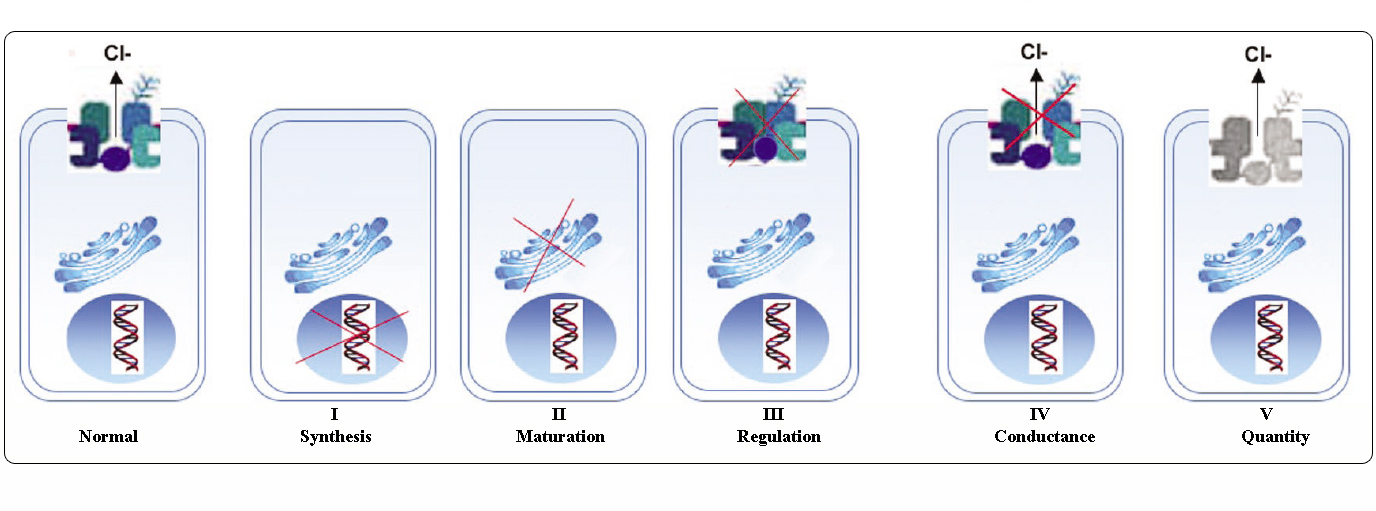
Adaptat de la Welsh și Smith (1993) și modificat de la Claustres (RBM online, 2005)
Mutațiile aparținând claselor I-III și VI conferă CFTR puțin sau deloc funcțional la nivelul membranei apicale și sunt considerate „severe” și conduc, de obicei, la un fenotip clasic de FC cu insuficiență pancreatică (CF-PI), deși severitatea bolii pulmonare poate fi variabilă.
Mutațiile aparținând claselor IV și V păstrează o anumită activitate reziduală a CFTR și conferă un fenotip mai blând. La pacienții cu cel puțin o alelă CF „ușoară”, funcția CFTR este de obicei suficientă pentru digestie, iar boala pulmonară este mai puțin severă. Indivizii cu mutanți CFTR care dau naștere la aproximativ 10% din nivelul normal de ARNm CFTR pot prezenta un singur simptom, cum ar fi CBAVD (absența bilaterală congenitală a vaselor deferente) la bărbați. Unele persoane heterozigote pentru o mutație CFTR ar putea prezenta un risc crescut de „tulburări legate de CFTR” (CFTR-RD), cum ar fi pancreatita, sinuzita sau aspergiloza bronhopulmonară alergică. În aceste cazuri, mutațiile CFTR ar putea fi un factor de predispoziție genetică, iar modificatorii genetici sau de mediu ar putea avea efecte adiționale.
Gruparea mutațiilor în diferite clase este utilă pentru înțelegerea mecanismului disfuncției, însă multe variații de secvență sunt rare/unice și datele disponibile nu permit evaluarea patogenității lor. Mai mult, o singură mutație poate cauza mai multe tipuri de anomalii sau poate conferi un fenotip variabil.
* Claustres M. 2005. Patologia moleculară a locusului CFTR în infertilitatea masculină. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Complexitatea într-o boală monogenică. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identificarea genei fibrozei chistice: analiză genetică. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL și alții. 1989. Identificarea genei fibrozei chistice: clonarea și caracterizarea ADN-ului complementar. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Localizarea fizică a doi markeri ADN strâns legați de locusul fibrozei chistice prin electroforeză pe gel în câmp pulsat. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. Mecanismele moleculare ale disfuncției canalului de clorură CFTR în fibroza chistică. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Secvența ADN-ului genomic al genei regulatorului de conductanță transmembranară a fibrozei chistice (CFTR). Genomics 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. Varianta genei CFTR pentru pacienții cu absența congenitală a vaselor deferente. Am J Hum Genet 57(4):958-60. PMID: 7573058.
.