 |
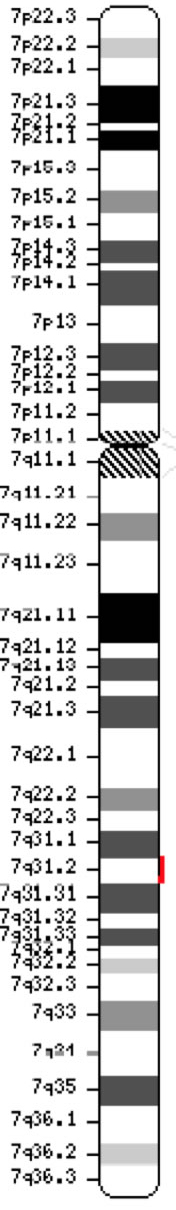
Genen CFTR (CFTR, OMIM #602421) är en medlem av ATP-bindande kassett underfamilj C (ABCC7). Genen, som upptäcktes 1989 (Kerem et al, 1989; Riordan et al, 1989; Rommens et al, 1989), sträcker sig över 188 702 bp på den långa armen av kromosom 7 (band 7q31-q32) (position 117 119 358-117 308 719) och består av 27 exoner som ursprungligen var numrerade 1-24 med underavdelningar a och b för exon 6, 14 och 17 (Zielenski et al., 1991). Det mogna transkriptet är 6 129 nukleotider långt inklusive en öppen läsram med 4 440 kodande baser (referenssekvens: NM_000492). Ensembl Gene ID: ENSG00000001626 Chromosom 7 ( NCBI, eller HGNC) |
Nomenklatur
Umd-CFTR använder sig av de rekommenderade nomenklaturriktlinjerna som föreslagits av Human Genome Variation Society (www.hgvs.org/mutnomen/) t.ex, cDNA-baserad numrering med A i ATG-translationsinitieringskodonet vid +1 och exoner numrerade från 1 till 27.
I UMD-CFTR är den kodande referenssekvensen NM_000492 med undantag för c.1408A i stället för G, och referensproteinsekvensen är NP_000483 med undantag för p.Met470 i stället för p.Val470.
För att underlätta för användare som är vana vid numreringen av konsortiet för mutationer i cystic fibrosis mutations database consortium (www.genet.sickkids.on.ca/cftr/) tillhandahålls två filer som kan laddas ner:
– en korrespondensnumrering för exoner och nukleotider: vanlig numrering (i vitt för exoner och i svart för nukleotider) och rekommenderad numrering (i rött). Den första och sista nukleotiden anges för varje exon. (ladda ner) (Visa/gömma)
– en korrespondenstabell för variationsnamn. (ladda ner)
Kodningssekvens (Visa/Hölje)
CFTR-mutationerna
Nästan 1 700 CFTR-sekvensmutationer och variationer har hittills beskrivits (www.genet.sickkids.on.ca/cftr). Den vanligaste mutationen i världen som är ansvarig för cystisk fibros är en deletion av tre baser som kodar för en fenylalaninrest i position 508, benämnd p.Phe508del, vilket försämrar CFTR-proteinets förmåga att veckla sig i det endoplasmatiska retikulumet (ER) och därmed ökar den snabba nedbrytningen av proteinet under ER-bearbetningen. Det står för cirka 70 % av CF-allelerna.
Heterologt uttryck av fullängds p.Phe508del-CFTR-cDNA har visat att detta mutantprotein endast förvärvar kärnglykosylering och inte komplexa oligosackaridkedjor och att det därför inte kan transporteras till cellytan, vilket stämmer överens med observationen av avsaknad av mutantprotein i färska epitelceller från patientvävnader. Bland andra skadliga effekter försämrar p.Phe508del den korrekta veckningen av CFTR och maskerar också konformativt den diacidiska utgångskoden för tre rester i NBD1 som tycks vara nödvändig för ER-export.
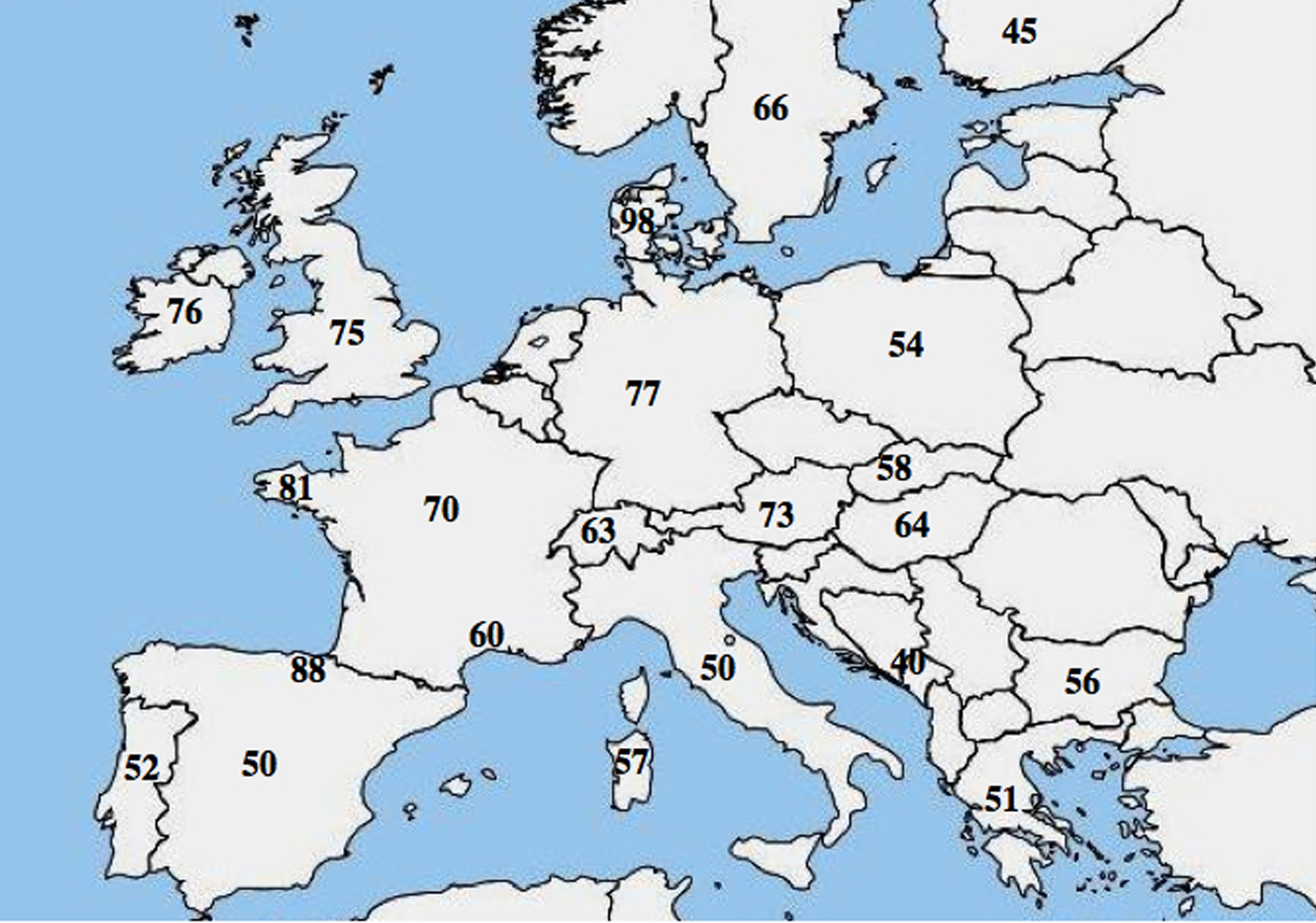
En molekylär allelisk heterogenitet i CF har rapporterats; ett flertal studier belyste en markant skillnad i frekvensen av p.Phe508del bland befolkningar och visade en minskande nordvästlig till sydöstlig gradient i Europa.
Klasser av CFTR-mutationer
Mutationer i CFTR-genen kan klassificeras i sex klasser beroende på deras molekylära mekanismer och konsekvenser för olika aspekter av CFTR:s biogenes, metabolism och funktion
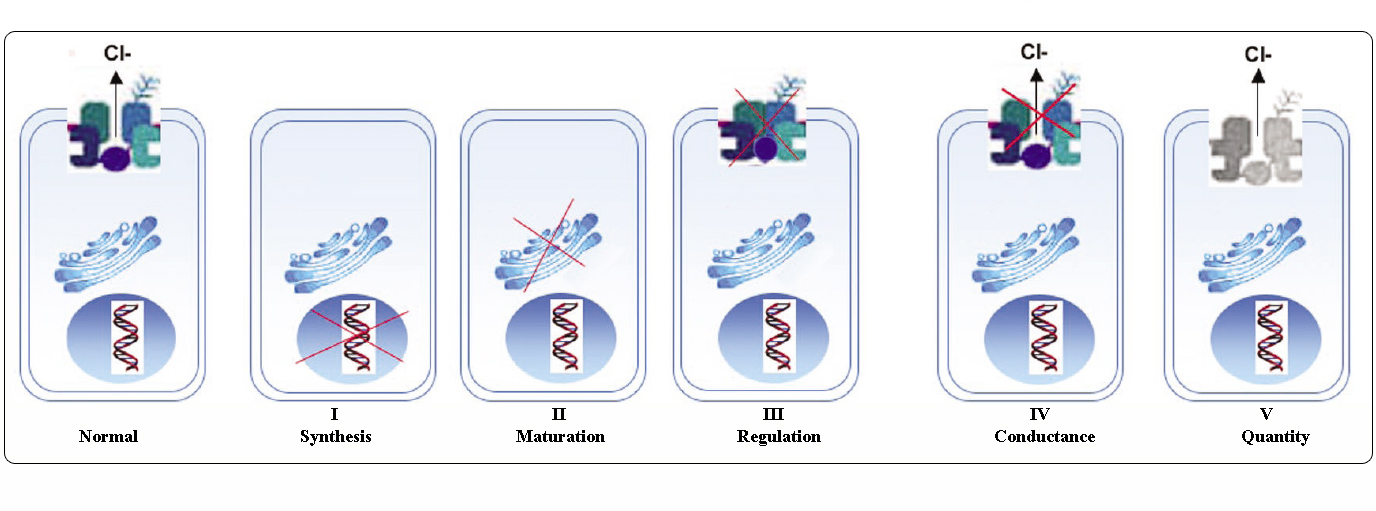
Adpterat från Welsh och Smith (1993) och modifierat från Claustres (RBM online, 2005)
Mutationer som tillhör klasserna I-III och VI ger liten eller ingen funktionell CFTR vid det apikala membranet och betraktas som ”allvarliga” och leder vanligen till en klassisk CF-fenotyp med bukspottskedsinsufficiens (CF-PI) även om lungsjukdomens allvarlighetsgrad kan vara varierande.
Mutationer som tillhör klasserna IV och V behåller en viss kvarvarande CFTR-aktivitet och ger en mildare fenotyp. Hos patienter med minst en ”mild” CF-allel är CFTR-funktionen vanligtvis tillräcklig för matsmältningen och lungsjukdomen är mindre allvarlig. Individer med CFTR-mutanter som ger upphov till cirka 10 % av den normala nivån av CFTR mRNA kan uppvisa endast ett symptom, t.ex. CBAVD (congenital bilateral absence of vas deferens) hos män. Vissa individer som är heterozygota för en CFTR-mutation kan ha en ökad risk för ”CFTR-relaterade sjukdomar” (CFTR-RD), t.ex. pankreatit, bihåleinflammation eller allergisk bronkopulmonell aspergillos. I dessa fall kan CFTR-mutationer vara en faktor för genetisk predisposition och genetiska eller miljörelaterade modifierande faktorer kan ha additiva effekter.
Gruppering av mutationer i olika klasser är användbart för att förstå dysfunktionens mekanism, men många sekvensvariationer är sällsynta/unikala och tillgängliga data gör det inte möjligt att bedöma deras patogenicitet. Dessutom kan en enskild mutation orsaka mer än en typ av avvikelse eller ge en varierande fenotyp.
* Claustres M. 2005. Molekylär patologi för CFTR-locus vid manlig infertilitet. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Komplexitet i en monogen sjukdom. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identifiering av genen för cystisk fibros: genetisk analys. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL m.fl. 1989. Identifiering av cystisk fibrosis-genen: kloning och karakterisering av komplementärt DNA. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Fysisk lokalisering av två DNA-markörer som är nära kopplade till cystisk fibrosis locus genom pulsad fältgelelektrofores. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. Molekylära mekanismer för CFTR-kloridkanalens dysfunktion vid cystisk fibros. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Genomisk DNA-sekvens för genen CFTR (cystic fibrosis transmembrane conductance regulator). Genomics 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. CFTR-genvariant för patienter med medfödd avsaknad av vas deferens. Am J Hum Genet 57(4):958-60. PMID: 7573058.