 |
El gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR, OMIM #602421) es un miembro de la subfamilia C de casetes de unión a ATP (ABCC7). El gen, descubierto en 1989 (Kerem et al, 1989; Riordan et al, 1989; Rommens et al, 1989), abarca 188.702 pb en el brazo largo del cromosoma 7 (banda 7q31-q32) (posición 117.119.358-117.308.719) y está compuesto por 27 exones numerados inicialmente del 1 al 24 con subdivisiones a y b para los exones 6, 14 y 17 (Zielenski et al., 1991). El transcrito maduro tiene una longitud de 6.129 nucleótidos, incluyendo un marco de lectura abierto de 4.440 bases codificantes (secuencia de referencia: NM_000492). Identificación de genes ennsembl: ENSG00000001626 Cromosoma 7 ( NCBI, o HGNC) |
Nomenclatura
El UMD-CFTR utiliza las directrices de nomenclatura recomendadas propuestas por la Human Genome Variation Society (www.hgvs.org/mutnomen/), por ejemplo numeración basada en el ADNc con la A del codón de iniciación traslacional ATG en +1 y los exones numerados del 1 al 27.
En el UMD-CFTR la secuencia de referencia de codificación es NM_000492 con la excepción de c.1408A en lugar de G, y la secuencia de proteína de referencia es NP_000483 con la excepción de p.Met470 en lugar de p.Val470.
Para comodidad de los usuarios que están acostumbrados a la numeración del consorcio de la base de datos de mutaciones de la fibrosis quística (www.genet.sickkids.on.ca/cftr/) se proporcionan dos archivos descargables:
– una numeración de correspondencia para exones y nucleótidos: numeración común (en blanco para los exones y en negro para los nucleótidos) y numeración recomendada (en rojo). Los primeros y últimos nucleótidos se indican para cada exón. (descargar) (Mostrar/Ocultar)
– una tabla de correspondencia para los nombres de las variaciones. (descargar)
Secuencia codificante (Mostrar/Ocultar)
Las mutaciones de CFTR
Hasta ahora se han descrito casi 1.700 mutaciones y variaciones de la secuencia de CFTR (www.genet.sickkids.on.ca/cftr). La mutación más común en todo el mundo responsable de la fibrosis quística es una deleción de tres bases que codifica un residuo de fenilalanina en la posición 508 denominada p.Phe508del, que perjudica la capacidad de la proteína CFTR para plegarse en el retículo endoplásmico (RE), potenciando así la rápida degradación de la proteína durante el procesamiento en el RE. Representa alrededor del 70% de los alelos de FQ.
La expresión heteróloga de la longitud completa de p.Phe508del-CFTR-ADN ha demostrado que esta proteína mutante adquiere sólo la glicosilación del núcleo y no las cadenas complejas de oligosacáridos y, como tal, no puede ser transportada a la superficie celular, de acuerdo con la observación de la ausencia de la proteína mutante en epitelios frescos de los tejidos de los pacientes. Entre otros efectos perjudiciales, p.Phe508del impide el correcto plegamiento de CFTR y también enmascara conformacionalmente el código de salida diácido de tres residuos en el NBD1 que parece necesario para la exportación al RE.
Se ha informado de una heterogeneidad alélica molecular en CF; numerosos estudios destacaron una marcada diferencia en la frecuencia de p.Phe508del entre las poblaciones y mostraron un gradiente decreciente del noroeste al sureste en Europa.
Clases de mutaciones de CFTR
Las mutaciones en el gen CFTR pueden clasificarse en seis clases según sus mecanismos moleculares y sus consecuencias para diferentes aspectos de la biogénesis, el metabolismo y la función de CFTR.
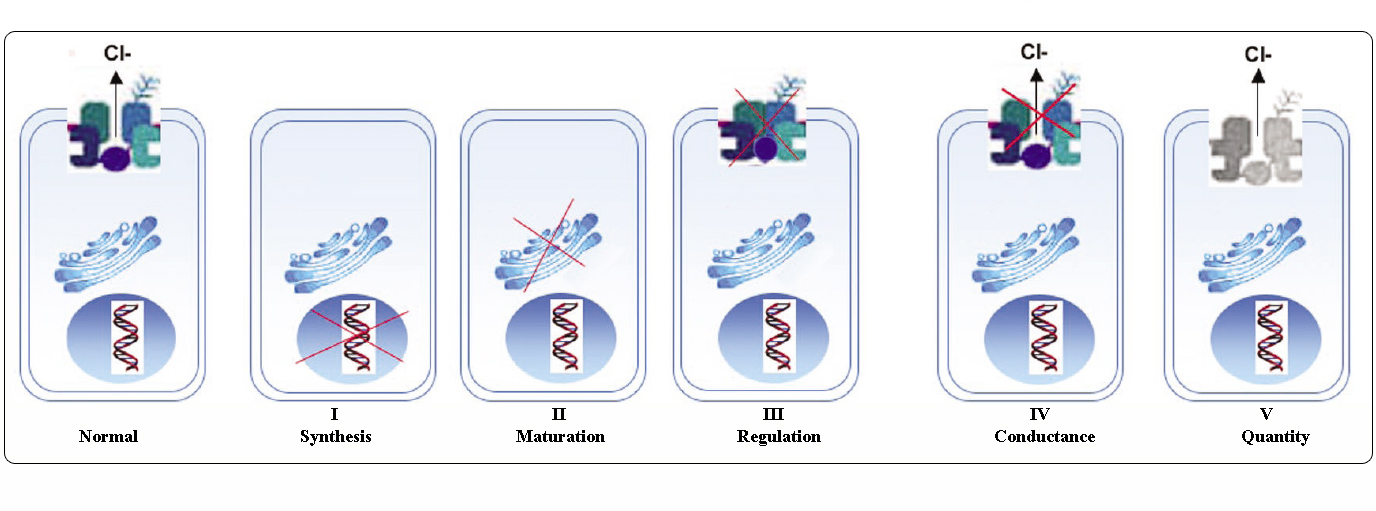
Adaptado de Welsh y Smith (1993) y modificado de Claustres (RBM online, 2005)
Las mutaciones pertenecientes a las clases I-III y VI confieren poco o ningún CFTR funcional en la membrana apical y se consideran como «severas» y suelen conducir a un fenotipo clásico de FQ con insuficiencia pancreática (CF-PI) aunque la gravedad de la enfermedad pulmonar puede ser variable.
Las mutaciones pertenecientes a las clases IV y V conservan cierta actividad residual de CFTR y confieren un fenotipo más leve. En los pacientes con al menos un alelo de FQ «leve», la función CFTR suele ser suficiente para la digestión y la enfermedad pulmonar es menos grave. Los individuos con mutantes de CFTR que dan lugar a un 10% del nivel normal de ARNm de CFTR pueden presentar sólo un síntoma, como la CBAVD (ausencia bilateral congénita de conductos deferentes) en los varones. Algunos individuos heterocigotos para una mutación de CFTR podrían tener un mayor riesgo de padecer «trastornos relacionados con CFTR» (CFTR-RD), como pancreatitis, sinusitis o aspergilosis broncopulmonar alérgica. En estos casos, las mutaciones de CFTR podrían ser un factor de predisposición genética y los modificadores genéticos o ambientales podrían desempeñar efectos aditivos.
La agrupación de las mutaciones en diferentes clases es útil para comprender el mecanismo de la disfunción, sin embargo, muchas variaciones de la secuencia son raras/únicas y los datos disponibles no permiten evaluar su patogenicidad. Además, una sola mutación puede causar más de un tipo de anomalía o puede conferir un fenotipo variable.
* Claustres M. 2005. Patología molecular del locus CFTR en la infertilidad masculina. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Complejidad en una enfermedad monogénica. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identificación del gen de la fibrosis quística: análisis genético. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL y otros. 1989. Identificación del gen de la fibrosis quística: clonación y caracterización del ADN complementario. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Localización física de dos marcadores de ADN estrechamente vinculados al locus de la fibrosis quística mediante electroforesis en gel de campo pulsado. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. Mecanismos moleculares de la disfunción del canal de cloruro CFTR en la fibrosis quística. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Secuencia del ADN genómico del gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR). Genómica 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. Variante del gen CFTR en pacientes con ausencia congénita de conductos deferentes. Am J Hum Genet 57(4):958-60. PMID: 7573058.